
INTRODUCCIÓN
Las RASopatías comprenden un grupo heterogéneo de síndromes genéticos causados por variantes en la línea germinal que afectan la vía de señalización de la quinasas activadas por mitógenos (Ras/MAPK) [1]. Esta cascada enzimática funciona como el centro de control para procesos biológicos tales como la proliferación celular, la diferenciación, la migración y la supervivencia [1,2]. Debido a que las mutaciones afectan puntos clave comunes de la misma vía, los síndromes resultantes—que incluyen Noonan, Cardio-Facio-Cutáneo (CFC) y Costello—comparten una notable superposición fenotípica que incluye características faciales dismórficas, anomalías cardiovasculares, defectos ectodérmicos y retraso en el desarrollo [1,3].
El síndrome de Costello (SC) representa uno de los fenotipos más severos y distintivos de este espectro. Fue descrito inicialmente en 1971 por el pediatra Jack Costello, quien observó un patrón único de retraso en el crecimiento, características faciales "toscas" y una laxitud cutánea inusual. Durante décadas, el diagnóstico dependió exclusivamente del reconocimiento clínico, lo que generó una confusión frecuente con el síndrome de Noonan. No fue hasta 2005 cuando Aoki y su equipo identificaron que las variantes de ganancia de función en el proto-oncogén HRAS eran responsables de esta entidad, permitiendo finalmente su distinción molecular definitiva.
A diferencia de las mutaciones somáticas altamente activadoras que se encuentran en varios tumores malignos, las variantes germinales presentes en las RASopatías son menos potentes, lo que permite la supervivencia del embrión durante la gestación, aunque a expensas de causar malformaciones multisistémicas [1,6]. En el CS, el gen HRAS actúa como un interruptor molecular que permanece "atrapado" en su forma activa debido a la incapacidad de hidrolizar GTP a GDP, saturando de forma permanente los efectores Raf, MEK y ERK [7]. Esta hiperactivación ininterrumpida es mucho más directa en el CS que en otras RASopatías donde el defecto es indirecto, lo que explica la mayor gravedad clínica y el 15% de riesgo de desarrollar cáncer que define esta patología [7,8]. El propósito de este informe es analizar las claves diagnósticas que permiten dirigir el estudio molecular hacia la variante HRAS como el eje del diagnóstico diferencial pediátrico.
PRESENTACIÓN DE CASO
Presentamos el caso de un recién nacido masculino, nacido de una madre primigrávida de 35 años con antecedentes de hipertensión arterial crónica. Durante la vigilancia prenatal, el estudio ecográfico reveló poliidramnios severo que requirió una amniocentesis reductora a las 34 semanas. El parto ocurrió a las 37 semanas por cesárea debido a sospecha de pérdida de bienestar fetal. El neonato presentó en estado crítico al nacer debido a asfixia perinatal severa. Se requirieron intubación endotraqueal inmediata y transferencia a la Unidad de Cuidados Intensivos Neonatales (UCIN).
En el examen físico inicial, el paciente presentó un fenotipo dismórfico llamativo: macrocefalia relativa con una frente prominente, hipertelorismo, orejas de implantación baja con hélices gruesas, fosas nasales antevertidas, y labios notablemente gruesos con una boca amplia. Un signo determinante fue la presencia de piel laxa y redundante, especialmente notable en la región nucal y extremidades, junto con pliegues profundos en las palmas y plantas, y hipoplasia ungueal generalizada. La evaluación abdominal identificó hepatomegalia palpable 5 cm por debajo del margen costal y criptorquidia bilateral.
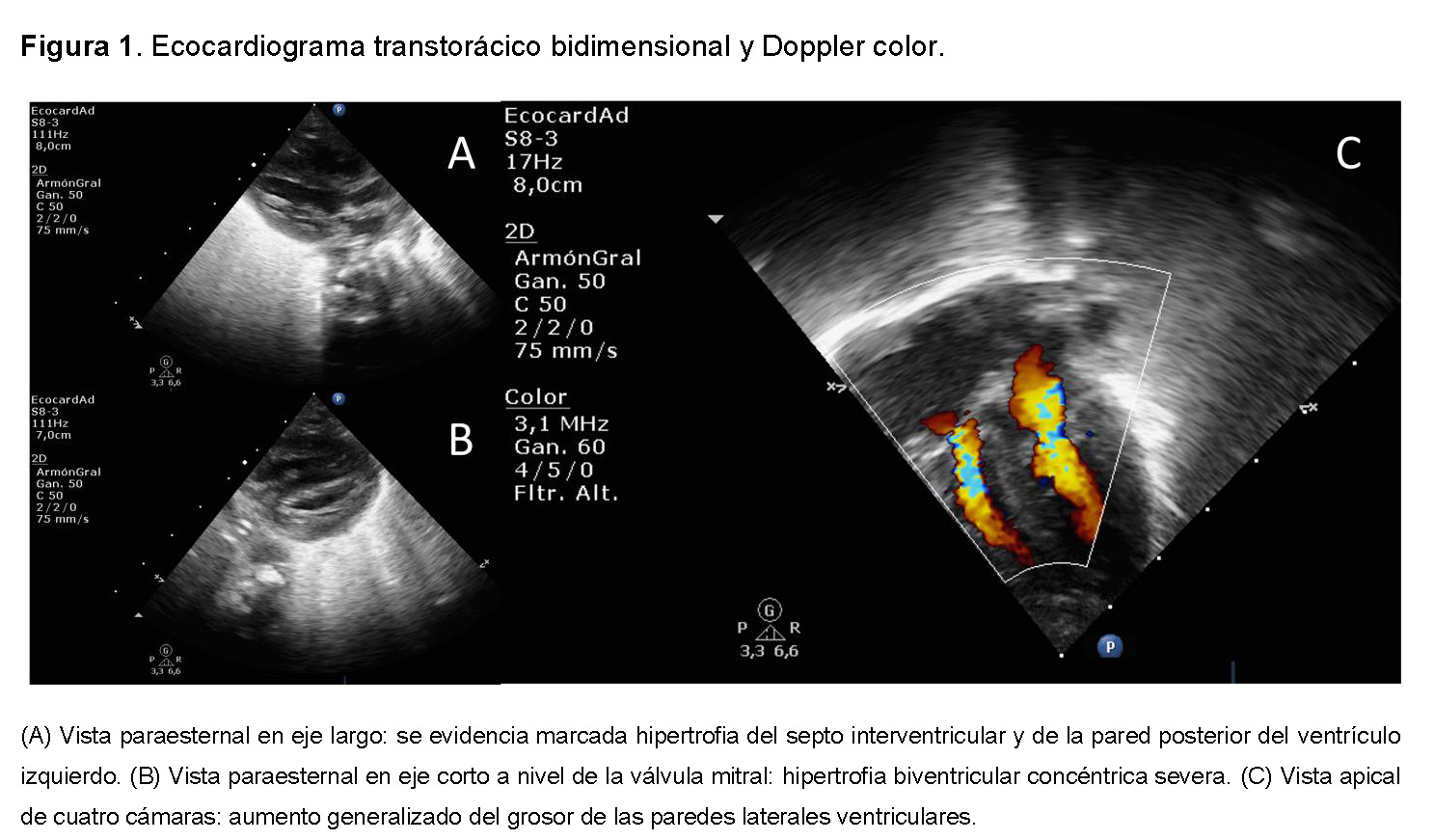
La evolución cardiovascular representó el mayor desafío terapéutico. Inicialmente, el ecocardiograma mostró leve hipertrofia septal; sin embargo, en evaluaciones seriadas, el paciente desarrolló miocardiopatía hipertrófica biventricular grave, concéntrica y progresiva (HCM) (Figura 1). Concomitantemente, el paciente presentó episodios de inestabilidad eléctrica caracterizados por taquiarritmias complejas, incluyendo flutter auricular y taquicardia supraventricular refractaria, lo que exigió el uso de terapia combinada con amiodarona y betabloqueantes.
Dada la fuerte sospecha de una RASopatía, se inició un enfoque genético escalonado. El microarreglo cromosómico inicial (CMA) no identificó variantes en el número de copias (CNV), reportando un resultado normal: arr(X,Y)x1, (1-22)x2. Dado que este resultado no descartaba mutaciones puntuales, se realizó secuenciación de todo el exoma (WES) utilizando la técnica CentoXome® Solo. El análisis genómico fragmentó enzimáticamente el ADN y lo enriqueció con sondas para 41 Mb del exoma codificante. Se logró una profundidad de lectura de al menos 20x en el 99.23% de las bases.
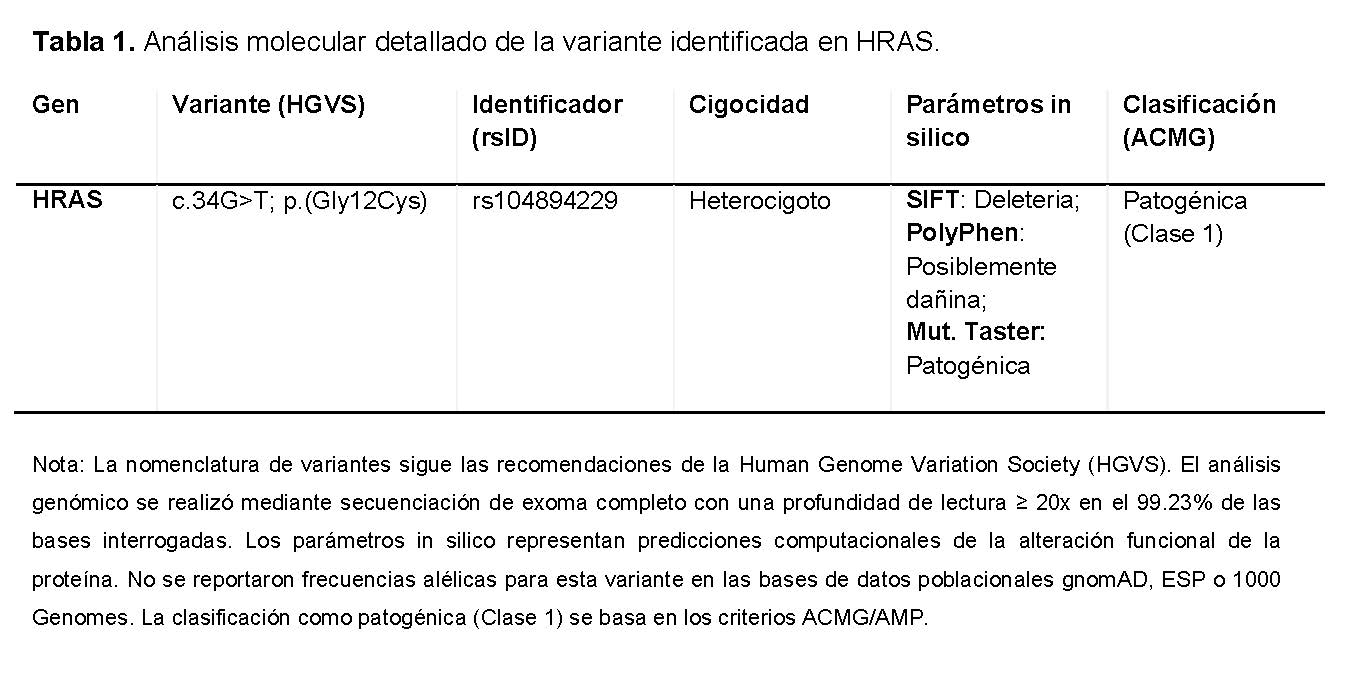
El estudio identificó una variante de un solo nucleótido en estado heterocigoto en el exon 2 del gen HRAS (NM_001130442.2:c.34G>T), que genera un cambio de aminoácido de Glicina a Cisteína en el codón 12 (p.Gly12Cys). Los predictores biotecnológicos (SIFT, PolyPhen, Mutation Taster) clasificaron unánimemente la variante como patogénica. Siguiendo las pautas del Colegio Americano de Genética Médica y Genómica (ACMG) y la Asociación de Patología Molecular (AMP), la variante fue clasificada como Clase 1 (Patogénica). El estudio parental confirmó que la variante era de novo, ratificando el diagnóstico definitivo del síndrome de Costello (OMIM 218040) (Tabla 1). La evolución intrahospitalaria fue extremadamente lenta. La insuficiencia respiratoria progresó a una dependencia absoluta del ventilador; los intentos de extubación fracasaron debido a una grave traqueomalacia que causaba un colapso dinámico de la vía aérea. Esto requirió una traqueostomía para asegurar la vía aérea y una gastrostomía para apoyo nutricional. El curso clínico se complicó con un neumotórax a tensión derecho y sepsis nosocomial por gérmenes multirresistentes. A pesar del apoyo multidisciplinario, el paciente murió por shock séptico y falla multiorgánica a los 5 meses de edad.
DISCUSIÓN
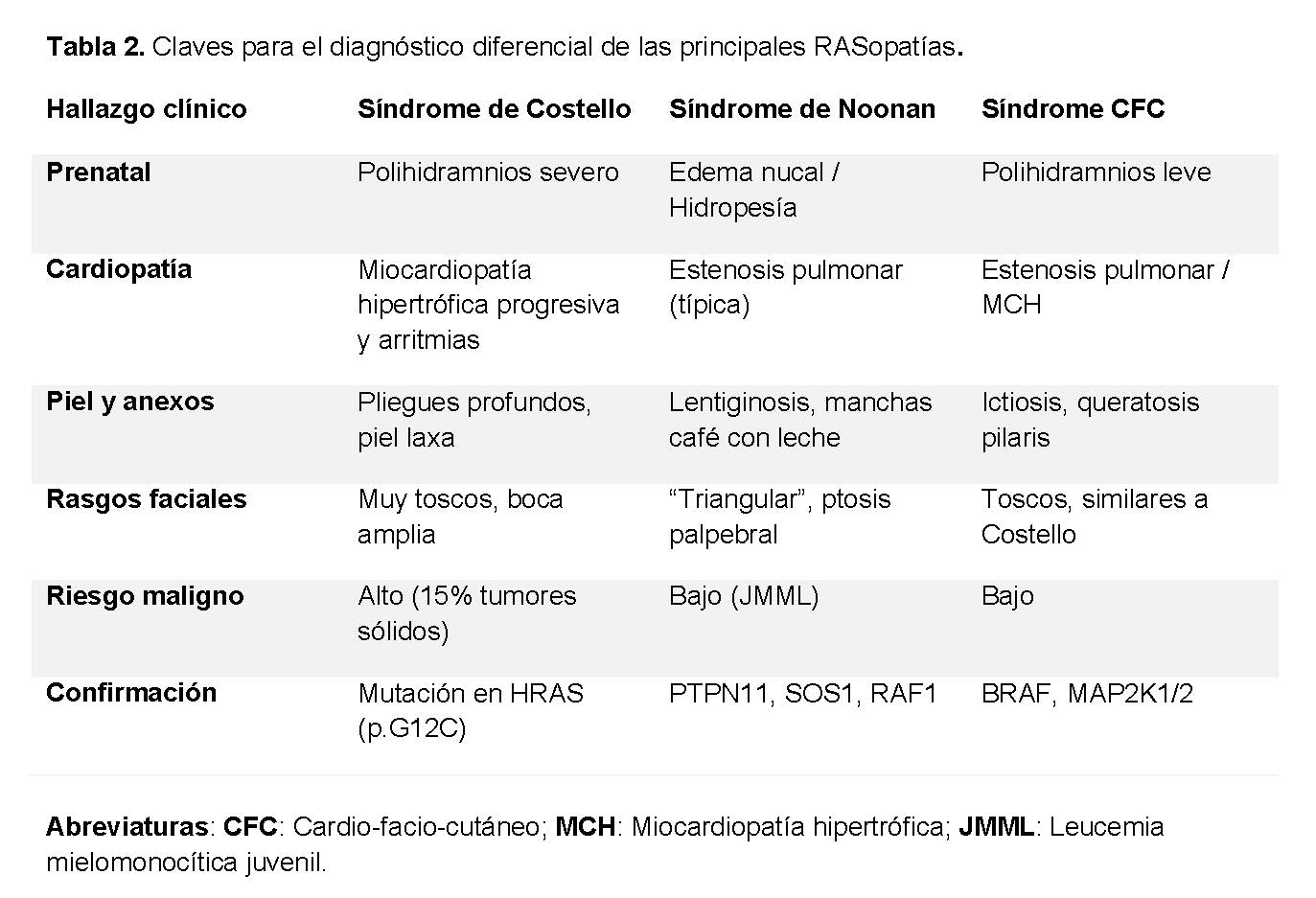
El diagnóstico diferencial de las RASopatías constituye uno de los mayores desafíos en la genética clínica pediátrica, dado el solapamiento de características y la variabilidad en la expresión neonatal. En la práctica clínica, la principal dificultad radica en diferenciar el síndrome de Costello (CS) del síndrome de Noonan y el síndrome Cardio-Facio-Cutáneo (CFC) (Tabla 2). Mientras que el síndrome de Noonan generalmente presenta un leve deterioro intelectual y una facies más triangular con ptosis palpebral, el CS se distingue por rasgos faciales notablemente gruesos y una discapacidad intelectual que generalmente varía de moderada a severa.
La sospecha clínica en nuestro caso se basó en el período gestacional. Según Lo et al., las manifestaciones neonatales extremas, como el polihidramnios severo y la hidropesía fetal, son indicativas de variantes HRAS de alto riesgo, diferenciándose del curso más benigno observado en otras patologías del espectro [9]. El polihidramnios en el CS refleja dificultades en la deglución fetal y un estado hipermetabólico que precede al fracaso en el crecimiento postnatal [3,4]. Al nacer, la extrema laxitud cutánea y los pliegues palmo-plantar profundos son marcadores externos de un desequilibrio proliferativo interno [3,7].
Una de las claves diagnósticas fundamentales observadas en este caso es la naturaleza del compromiso cardiovascular. Coincidiendo con Lin et al., aunque la estenosis de la válvula pulmonar es la lesión más frecuente en el síndrome de Noonan, en el CS, la miocardiopatía hipertrófica (MCH) está presente en el 61% de los pacientes y puede ser rápidamente progresiva en los dos primeros años de vida [10]. Además, la presencia de taquicardia atrial multifocal o flutter auricular se identifica como un marcador de alta especificidad para el CS, facilitando el diagnóstico diferencial temprano de otras causas de hipertrofia cardíaca [10,11].
La agresividad clínica del paciente es consistente con la literatura que asocia la variante p.Gly12Cys con presentaciones severas. A diferencia de la variante más común (p.G12S, responsable del 80-90% de los casos y con un riesgo oncológico del 7%), la variante p.Gly12Cys está vinculada a un remodelado miocárdico acelerado desde el nacimiento y a una dificultad respiratoria persistente [9,12]. Bertola et al. han informado que incluso variantes en el codón 13 (como p.G13D) pueden dar lugar a fenotipos más atenuados, destacando la importancia de la correlación genotipo-fenotipo para establecer el pronóstico [13].
Un aspecto crítico del diagnóstico es el riesgo neoplásico
CS tiene un riesgo acumulativo de por vida del 15% de desarrollar tumores malignos, principalmente rabdomiosarcoma embrional, neuroblastoma y carcinoma de vejiga [3,4]. Estep et al. demostraron que la progresión hacia la malignidad generalmente sigue un modelo de "segundo golpe", donde el alelo silvestre se pierde (LOH) en el tejido tumoral, lo que aumenta el crecimiento celular descontrolado [6]. Por lo tanto, la confirmación de la mutación HRAS requiere un protocolo de vigilancia estricto: ecografía abdominal y pélvica cada 3-6 meses hasta los 8-10 años de edad, y análisis de catecolaminas o citología urinaria dependiendo de la edad del paciente [8].
Desde una perspectiva técnica, el microarreglo negativo en este paciente es un hallazgo esperado, ya que las RASopatías son causadas por mutaciones puntuales y no por desequilibrios cromosómicos estructurales. Esto ratifica la secuenciación de exoma completo (WES) como el estándar de oro actual [5,12]. Aunque el enfoque en nuestro caso fue paliativo debido a complicaciones sistémicas, la precisión diagnóstica abre la puerta a futuras intervenciones. Hebron et al. describen el uso emergente de inhibidores de MEK (como el trametinib) para reducir la masa ventricular en pacientes con HCM severa, ofreciendo una alternativa terapéutica no quirúrgica en investigación [14].
Hasta donde sabemos, este constituye el primer caso documentado de síndrome de Costello con confirmación diagnóstica mediante secuenciación de exomas completos reportado en Ecuador, subrayando la relevancia de la genómica clínica en el abordaje de enfermedades raras en nuestro país.
CONCLUSIÓN
La confirmación diagnóstica del síndrome de Costello mediante WES representa un punto de inflexión fundamental en el enfoque de las RASopatías neonatales. El síndrome de Costello no solo es un síndrome dismorfico, sino una condición premaligna y cardiovascularmente agresiva que requiere una gestión multidisciplinaria coordinada entre genética, cardiología y oncología.
El diagnóstico genético temprano es indispensable para establecer protocolos de vigilancia anticipatoria y para proporcionar asesoramiento genético preciso a la familia. En este caso, encontrar una variante de novo ayudó a mitigar la incertidumbre sobre la recurrencia en futuros embarazos. La integración de la genómica en la práctica pediátrica es esencial para transformar la sospecha clínica en medicina de precisión, permitiendo incluso considerar nuevas estrategias farmacológicas que prometen cambiar la historia natural de estas enfermedades ultra-raras.
Contribuciones de los autores
AGC: Conceptualización, Investigación, Curaduría de datos y Redacción - borrador original. LMP: Supervisión, Validación y Redacción - revisión y edición. RERP: Investigación (evaluación ecocardiográfica y procesamiento de imágenes), Validación y Visualización. Todos los autores contribuyeron significativamente a la concepción, diseño, recolección de datos clínicos, análisis genético y revisión crítica del contenido intelectual de este manuscrito.
Abreviaturas
ACMG: Colegio Americano de Genética Médica y Genómica; AMP: Asociación para la Patología Molecular; CFC: Cardio-Facio-Cutáneo; CMA: Microarreglo cromosómico; CNV: Variantes en el número de copias; CS: Síndrome de Costello; GDP/GTP: Difosfato de guanosina / Trifosfato de guanosina; HCM: Miocardiopatía hipertrófica; HGVS: Sociedad de Variación del Genoma Humano; JMML: Leucemia Mieloide Juvenil; MAPK: Quinasas activadas por mitógenos; NICU: Unidad de Cuidados Intensivos Neonatales; y WES: Secuenciación de exomas completos.
Perspectiva Familiar
A pesar del desenlace fatal, los padres del paciente expresaron su disposición a compartir este caso con la comunidad médica. Su principal motivación es contribuir al conocimiento científico del Síndrome de Costello, esperando que la difusión de estos hallazgos facilite el diagnóstico temprano y mejore el manejo de futuros pacientes.