
INTRODUCTION
RASopathies comprise a heterogeneous group of genetic syndromes caused by germline variants that affect the mitogen-activated protein kinase (Ras/MAPK) signaling pathway [1]. This enzymatic cascade functions as the control center for biological processes such as cell proliferation, differentiation, migration, and survival [1,2]. Because mutations affect common key points of the same pathway, the resulting syndromes—which include Noonan, Cardio-Facio-Cutaneous (CFC), and Costello—share a notable phenotypic overlap that includes dysmorphic facial features, cardiovascular anomalies, ectodermal defects, and developmental delay [1,3].
Costello syndrome (CS) represents one of the most severe and distinctive phenotypes of this spectrum. It was initially described in 1971 by pediatrician Jack Costello, who observed a unique pattern of growth retardation, "coarse" facial features, and unusual skin laxity [3,4]. For decades, diagnosis depended exclusively on clinical recognition, which generated frequent confusion with Noonan syndrome. It was not until 2005 when Aoki and his team identified that gain-of-function variants in the HRAS proto-oncogene were responsible for this entity, finally allowing its definitive molecular distinction [5].
nlike the highly activating somatic mutations found in various malignant tumors, the germline variants present in RASopathies are less potent, which allows embryo survival during gestation, albeit at the expense of causing multisystemic malformations [1,6]. In CS, the HRAS gene acts as a molecular switch that remains "trapped" in its active form due to the inability to hydrolyze GTP to GDP, permanently saturating the Raf, MEK, and ERK effectors [7]. This uninterrupted hyperactivation is much more direct in CS than in other RASopathies where the defect is indirect, which explains the greater clinical severity and the 15% risk of developing cancer that defines this pathology [7,8]. The purpose of this report is to analyze the diagnostic keys that allow the molecular study to be directed toward the HRAS variant as the axis of pediatric differential diagnosis.
CASE PRESENTATION
We present the case of a male newborn, born to a 35-year-old primigravid mother with a history of chronic arterial hypertension. During prenatal surveillance, the ultrasound study revealed severe polyhydramnios that required a reductive amniocentesis at 34 weeks. Delivery occurred at 37 weeks by cesarean section due to suspected loss of fetal well-being. The neonate presented in critical condition at birth due to severe perinatal asphyxia. Immediate endotracheal intubation and transfer to the Neonatal Intensive Care Unit (NICU) were required.
On initial physical examination, the patient exhibited a striking dysmorphic phenotype: relative macrocephaly with a prominent forehead, hypertelorism, low-set ears with thick helices, anteverted nostrils, and markedly thickened lips with a wide mouth. A determining sign was the presence of lax and redundant skin, especially notable in the nuchal region and extremities, along with deep creases on the palms and soles, and generalized nail hypoplasia. The abdominal evaluation identified palpable hepatomegaly 5 cm below the costal margin and bilateral cryptorchidism.
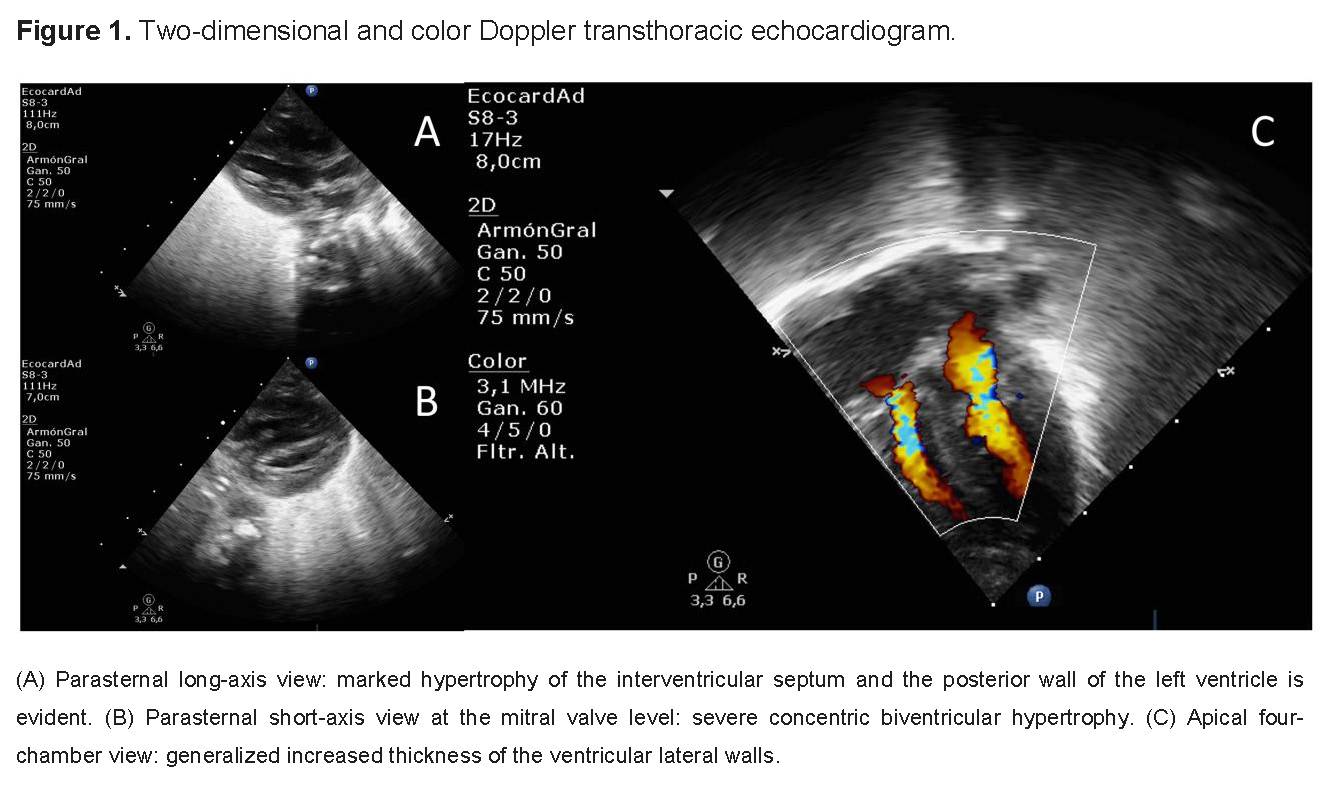
The cardiovascular evolution represented the greatest therapeutic challenge. Initially, the echocardiogram showed mild septal hypertrophy; however, in serial evaluations, the patient developed severe, concentric, and progressive biventricular hypertrophic cardiomyopathy (HCM) (Figure 1). Concomitantly, the patient presented episodes of electrical instability characterized by complex tachyarrhythmias, including atrial flutter and refractory supraventricular tachycardia, which mandated the use of combined therapy with amiodarone and beta-blockers.
Given the strong suspicion of a RASopathy, a stepped genetic approach was initiated. The initial chromosomal microarray (CMA) did not identify copy number variants (CNV), reporting a normal result: arr(X,Y)x1, (1-22)x2. Since this result did not rule out point mutations, whole-exome sequencing (WES) was performed using the CentoXome® Solo technique. The genomic analysis enzymatically fragmented the DNA and enriched it with probes for 41 Mb of the coding exome. A read depth of at least 20x was achieved in 99.23% of the bases.
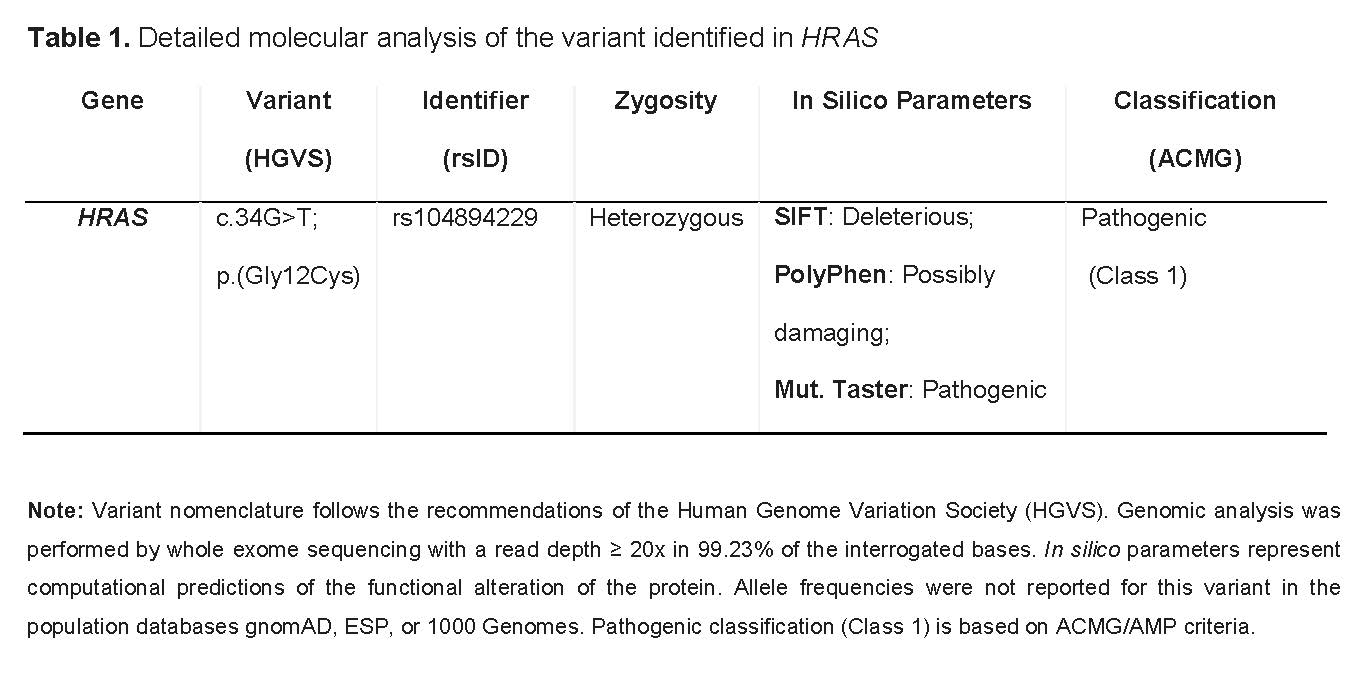
The study identified a single-nucleotide variant in a heterozygous state in exon 2 of the HRAS gene (NM_001130442.2:c.34G>T), which generates an amino acid change of Glycine to Cysteine at codon 12 (p.Gly12Cys). Biotechnological predictors (SIFT, PolyPhen, Mutation Taster) unanimously rated the variant as pathogenic. Following the guidelines of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP), the variant was classified as Class 1 (Pathogenic). The parental study confirmed that the variant was de novo, ratifying the definitive diagnosis of Costello syndrome (OMIM 218040) (Table 1).
The in-hospital evolution was extremely torpid. Respiratory failure progressed to absolute ventilator dependence; extubation attempts failed due to severe tracheomalacia causing dynamic airway collapse. This necessitated a tracheostomy to secure the airway and a gastrostomy for nutritional support. The clinical course was complicated by a right tension pneumothorax and nosocomial sepsis by multiresistant germs. Despite multidisciplinary support, the patient died from septic shock and multiorgan failure at 5 months of age.
DISCUSSION
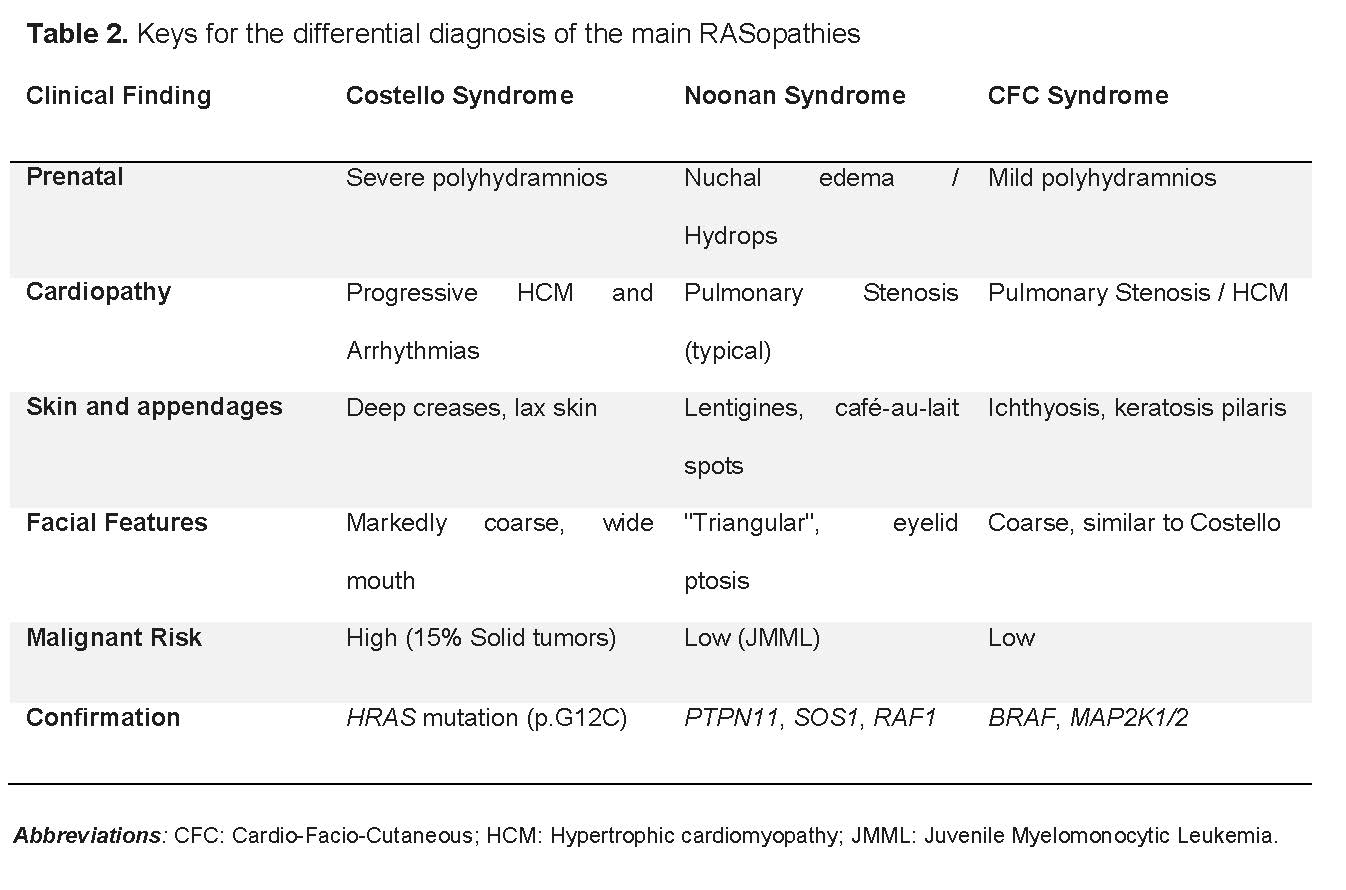
The differential diagnosis of RASopathies constitutes one of the greatest challenges in pediatric clinical genetics, given the overlap of features and the variability in neonatal expression. In clinical practice, the main difficulty lies in differentiating CS from Noonan syndrome and Cardio-Facio-Cutaneous (CFC) syndrome (Table 2). While Noonan syndrome usually presents mild intellectual impairment and a more triangular facies with palpebral ptosis, CS is distinguished by markedly coarse facial features and an intellectual disability that usually ranges from moderate to severe [2,4].
Clinical suspicion in our case was based from the gestational period. According to Lo et al., extreme neonatal manifestations such as severe polyhydramnios and fetal hydrops are indicative of high-risk HRAS variants, differentiating from the more benign course observed in other pathologies of the spectrum [9]. Polyhydramnios in CS reflects difficulties in fetal swallowing and a hypermetabolic state that precedes postnatal failure to thrive [3,4]. At birth, extreme skin laxity and deep palmar-plantar creases are external markers of an internal proliferative imbalance [3,7].
One of the fundamental diagnostic keys observed in this case is the nature of the cardiovascular involvement. Coinciding with Lin et al., while pulmonary valve stenosis is the most frequent lesion in Noonan syndrome, in CS, hypertrophic cardiomyopathy (HCM) is present in 61% of patients and can be rapidly progressive in the first two years of life [10]. Furthermore, the presence of multifocal atrial tachycardia or atrial flutter is identified as a marker of high specificity for CS, facilitating early differential diagnosis from other causes of cardiac hypertrophy [10,11].
The clinical aggressiveness of the patient is consistent with the literature associating the p.Gly12Cys variant with severe presentations. Unlike the most common variant (p.G12S, responsible for 80-90% of cases and with a 7% oncological risk), the p.Gly12Cys variant is linked to accelerated myocardial remodeling from birth and persistent respiratory distress [9,12]. Bertola et al. have reported that even variants in codon 13 (such as p.G13D) can yield more attenuated phenotypes, highlighting the importance of genotype-phenotype correlation for establishing prognosis [13].
A critical aspect of the diagnosis is the neoplastic risk. CS carries a lifetime cumulative risk of 15% for developing malignant tumors, primarily embryonal rhabdomyosarcoma, neuroblastoma, and bladder carcinoma [3,4]. Estep et al. demonstrated that progression toward malignancy usually follows a "second hit" model, where the wild-type allele is lost (LOH) in the tumor tissue, enhancing uncontrolled cell growth [6]. Therefore, confirmation of the HRAS mutation mandates a strict surveillance protocol: abdominal and pelvic ultrasound every 3-6 months until 8-10 years of age, and catecholamine analysis or urinary cytology depending on the patient's age [8].
From a technical perspective, the negative microarray in this patient is an expected finding, as RASopathies are caused by point mutations and not by structural chromosomal imbalances. This ratifies whole-exome sequencing (WES) as the current gold standard [5,12]. Although the approach in our case was palliative due to systemic complications, diagnostic precision opens the door to future interventions. Hebron et al. describe the emerging use of MEK inhibitors (such as trametinib) to reduce ventricular mass in patients with severe HCM, offering a non-surgical therapeutic alternative under investigation [14].
To the best of our knowledge, this constitutes the first documented case of Costello syndrome with diagnostic confirmation by whole-exome sequencing reported in Ecuador, highlighting the relevance of clinical genomics in the approach to rare diseases in our country.
CONCLUSION
The diagnostic confirmation of Costello syndrome by WES represents a fundamental turning point in the approach to neonatal RASopathies. CS is not only a dysmorphic syndrome but a premalignant and cardiovascularly aggressive condition that requires coordinated multidisciplinary management among genetics, cardiology, and oncology.
Early genetic diagnosis is indispensable for establishing anticipatory surveillance protocols and for providing precise genetic counseling to the family. In this case, finding a de novo variant helped mitigate uncertainty about recurrence in future pregnancies. The integration of genomics into pediatric practice is essential to transform clinical suspicion into precision medicine, even allowing consideration of new pharmacological strategies that promise to change the natural history of these ultra-rare diseases.
Author Contributions
AGC: Conceptualization, Investigation, Data curation, and Writing – original draft. LMP: Supervision, Validation, and Writing – review & editing. RERP: Investigation (echocardiography assessment and image processing), Validation, and Visualization. All authors contributed significantly to the conception, design, clinical data collection, genetic analysis, and critical review of the intellectual content of this manuscript.
Abbreviations
ACMG: American College of Medical Genetics and Genomics; AMP: Association for Molecular Pathology; CFC: Cardio-Facio-Cutaneous; CMA: Chromosomal microarray; CNV: Copy number variants; CS: Costello syndrome; GDP/GTP: Guanosine diphosphate / Guanosine triphosphate; HCM: Hypertrophic cardiomyopathy; HGVS: Human Genome Variation Society; JMML: Juvenile Myelomonocytic Leukemia; MAPK: Mitogen-activated protein kinase; NICU: Neonatal Intensive Care Unit; and WES: Whole-exome sequencing.
Family Perspective
Despite the fatal outcome, the patient's parents expressed their willingness to share this case with the medical community. Their main motivation is to contribute to the scientific knowledge of Costello Syndrome, hoping that the dissemination of these findings facilitates early diagnosis and improves the management of future patients.