
Agrecanopatías: Reporte de Displasia Espóndiloepifisiaria tipo Kimberley (SEDK) en una familia, producida por variante probablemente patogénica del gen ACAN no descrita previamente
Aggrecanopathies: Report of a Spondyloepiphyseal Dysplasia Kimberley type (SEDK) in a family, caused by a previously undescribed likely pathogenic variant of the ACAN geneEnrique Daniel Austin-Ward1
 , Jean Villegas2
, Jean Villegas21. Instituto INDICASAT (Instituto de Investigaciones Científicas Avanzadas y Servicios de Alta Tecnología), Panamá, Rep. de Panamá;Hospital Pacífica Salud, Panamá, República de Panamá;Instituto de Genética Médica y Genómica, Caja de Seguro Social, Panamá, Rep. de Panamá; 2. Consultorio Pediátrico Villegas Díaz, Panamá, Rep. de Panamá.
Descargas
Resumen
El estudio de pacientes con talla baja es complejo y requiere de algoritmos diagnósticos en los cuales la evaluación por parte de Genética es esencial, buscando descartar síndromes genéticos que puedan explicar las manifestaciones del paciente. Entre estas posibles causas, las displasias esqueléticas deben ser tomadas en todo momento como una posibilidad diagnóstica. Presentamos el caso de una familia en la cual en el probando y en su progenitor se detectó una variante probablemente patogénica no descrita previamente en el gen ACAN el cual codifica el Proteoglicano (PG) Aggrecan, componente fundamental en el crecimiento óseo endocondral y en el cartílago articular, y cuya alteración en estado de heterocigoto produce la Displasia Esquelética Espóndiloepifisiaria tipo Kimberley (SEDK) y otros trastornos conocidos como aggrecanopatías. Los análisis genéticos moleculares se convierten en estudios mandatorios para lograr el diagnóstico acertado en estos casos. Se espera que el estudio de estas condiciones dé luces para la mejor comprensión de la fisiopatología de la Osteoartritis y para el desarrollo de nuevos tratamientos para su manejo, dada su elevada prevalencia en adultos mayores y a que en ella la degeneración del aggrecano juega un rol central.
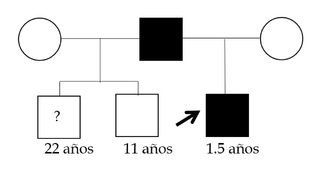
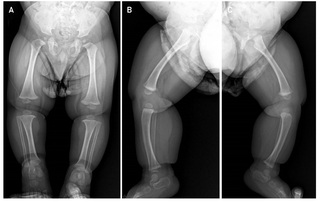
Se trata de infante masculino de 2 años, primer hijo de padres no consanguíneos, sin antecedentes relevantes del embarazo y del parto a término y sin complicaciones, quien es referido a la consulta de Genética por su médico pediatra por talla baja severa (- 5DS), y con un proceso de sospecha diagnóstica en curso de una displasia esquelética. El paciente tiene dos medios hermanos paternos (Figura 1). Al examen físico el paciente presentaba hallazgos dismorfológicos leves que incluían hipoplasia de la parte media de la cara, puente nasal plano, macrocefalia relativa, protuberancia frontal, braquidactilia y lordosis (Figura 2). Evaluaciones radiográficas a los 11 meses de todo el esqueleto revelaron como único hallazgo notable ensanchamiento en copa de ambos fémures con discreto acortamiento longitudinal de ambas tibias con curvatura anteroposterior sugerente de displasia ósea (Figura 3), y con un estudio edad ósea, que revelaba una edad ósea avanzada para la edad de 1 año y 3 meses (4 meses). El paciente recibió estimulación temprana por haberse detectado un ligero retraso del desarrollo psicomotor con hipertonía y al año ya cumplía con todos los hitos del desarrollo. En el proceso de evaluación se realizaron pruebas genéticas moleculares.

Pruebas Moleculares
Se realizó un estudio de Secuenciación de Genoma Completo (WGS) con Análisis Multiómico (Centogenome ® MOx 2.0) en Centogene, que involucra también el estudio transcriptómico. El ADN genómico fue fragmentado enzimáticamente y marcado con secuencias adaptadoras compatibles con Ilumina. Las librerías fueron secuenciadas desde ambos extremos (paired-end) en una plataforma Illumina para generar una profundidad de cobertura media de ~ 30x. Se aplicó un proceso bioinformático basado en el proceso DRAGEN de Illumina, así como el proceso bioinformático interno de CENTOGENE. Las lecturas fueron alineadas con el ensamblaje del genoma Genome Reference Consortium Human Build 37 (GRCh37/hg19), así como a la secuencia de referencia de Cambridge (revised Cambridge Referece Sequence; rCRS) del ADN mitocondrial humano (NC_012920). Las variantes de secuencia (SNVs/indels) y las variantes en el número de copia (CNVs) fueron llamadas usando el algoritmo DRAGEN, Manta y propio. Se evaluaron todas las variantes con una frecuencia alélica minoritaria (MAF) inferior al 1% en la base de datos gnomAD, y las variantes causantes de enfermedad reportadas en HGMD®, en ClinVar o en el Biodatabank de CENTOGENE. Aunque la evaluación se centró en los exones codificantes y las regiones intrónicas flanqueantes, el gen completo se interrogó en busca de variantes candidatas con una asociación plausible con el fenotipo. Se consideraron todos los patrones potenciales de modo de herencia. Además, se utilizaron los antecedentes familiares y la información clínica proporcionada para evaluar las variantes identificadas con respecto a su patogenicidad y causalidad de la enfermedad. Las variantes se clasifican en cinco clases (patogénica, probablemente patogénica, VUS, probablemente benigna y benigna) siguiendo las directrices del ACMG para la clasificación de variantes además de las recomendaciones de ClinGen. Se informaron todas las variantes relevantes relacionadas con el fenotipo del paciente. Se aisló el ARN a partir de CentoCard®. El ARN mensajero (ARNm) se enriqueció, fragmentó y transcribió en ADN complementario (ADNc). A continuación, se realizó la ligación de los adaptadores y la amplificación de las librerías, que se secuenciaron por pares en una plataforma Illumina. Los datos de secuenciación se procesaron mediante un pipeline bioinformático propio. Las lecturas de secuenciación se evaluarón mediante herramientas de control de calidad con el fin de garantizar la integridad de los datos e identificar posibles problemas (lecturas de baja calidad, contaminación por adaptadores, etc.). Posteriormente, las lecturas se alinearon frente al genoma de referencia GRCh37/hg19 mediante STAR, que permite determinar con precisión los eventos de splicing. Se aplicaron criterios de calidad para aceptar o rechazar los datos de secuenciación. Los datos aceptados se analizaron y se utilizaron como pruebas adicionales para evaluar el efecto de la variante en el proceso de splicing.
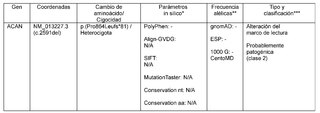
Se detectó una variante en el gen ACAN, consistente en c.2591del p.(Pro864Leufs*81) en estado de heterocigoto, que causa un cambio en el marco de lectura a partir del codón 864. El nuevo marco de lectura termina en un codón de parada 80 posiciones adelante. Se clasifica como probablemente patogénica (clase 2) de acuerdo con las recomendaciones de CENTOGENE y la ACMG (Tabla 1). Las variantes patogénicas en el gen ACAN se asocian a displasia espondiloepifisaria tipo Kimberley (SEDK) y baja estatura y edad ósea avanzada, con o sin osteoartritis y/o osteocondritis disecante de aparición temprana (SSOAOD).
Obtenidos estos resultados y en la discusión con los padres del paciente emergieron datos no comentados previamente relacionados con el padre y uno de los medios hermanos del paciente. El padre tuvo a los 43 años, una cirugía por hernia de disco y el medio hermano mayor, padecimientos en una de sus rodillas con ruptura y cambios degenerativos en sus meniscos. Basados en estos datos y en la indicación del proceso diagnóstico, se procedió a realizar el estudio de secuenciación del gen ACAN en los padres, detectándose la misma mutación en el padre del probando. Al momento de escribir el artículo no había sido posible investigar a los medios hermanos del paciente.
La integridad del desarrollo y funcionamiento de los elementos de sostén del organismo entre los que se encuentran los del sistema esquelético y la matriz extracelular, depende a su vez de la integridad molecular de sus componentes. Alteraciones genéticas en los mismos pueden producir anomalías en la estructura y en la talla de los individuos.
Entre estos elementos estructurales moleculares se encuentran los glicosoaminoglicanos y los proteoglicanos. Los glicosaminoglicanos (GAGs) son polisacáridos de origen mesodérmico, propios de los tejidos animales y consisten en heteropolisacáridos constituidos por carbohidratos complejos de gran tamaño no ramificados compuestos por unidades de disacáridos repetidas. Los Proteoglicanos (PG), por otro lado, tienen una estructura formada por glicosaminoglicanos unidos a un esqueleto central de proteína o “core” [1].
Los PG son moléculas muy variables, ubicuas y con múltiples roles en el organismo aparte del rol estructural ya mencionado, y que incluyen división celular, adhesión, extensión, migración, quimioatracción, guía de axones, ensamblaje de matriz extracelular, captación de lipoproteínas, proteólisis e ingreso de los virus a las células e interactúan con muchas moléculas tales comofactores de crecimiento, proteínas de unión a factores de crecimiento, proteasas extracelulares, inhibidores de proteasas, moléculas de la matriz extracelular y son capaces de controlar gradientes y disponibilidad de citoquinas, quemoquinas y morfógenos entre otras moléculas y en diversas circunstancias y entornos celulares, como los involucrados en homeostasis, desarrollo embrionario, reparación y en el mecanismode enfermedades como el cáncer [1,2]. A pesar de esta gran diversidad de ubicaciones y roles, comprenden el número relativamente pequeño de 50 genes relacionados al esqueleto central de proteína [3]. Una de las diversas formas de clasificar a los PG consiste en dividirlos en familias en base a su localización predominante, resultando cuatro clases principales (intracelular, superficie celular, pericelular y extracelular) [4].
Alteraciones genéticas en los PG se han asociado a distintas condiciones, aparte de diversos tipos de cáncer, y que incluyen la Enfermedad de Alzheimer, desórdenes relacionados al amiloide, implicaciones en procesos infecciosos como el relacionado al SARS-CoV-2, ateroesclerosis y el Síndrome de Ehlers-Danlos entre otras [3,5].
Muy relacionado con el Síndrome de Ehlers-Danlos y sus muy diversas manifestaciones, están las relacionadas a otros defectos de los PG en la matriz extracelular con un efecto marcadamente estructural y que producen alteraciones del esqueleto. Dentro de las familias de PG que tienen predominio extracelular, se encuentra a su vez la familia Hyalectan/Lectican que incluye cuatro genes: ACAN, VCAN, NCAN y BCAN, correspondientes a los PG aggrecan, versican, neurocan y brevican respectivamente [4].
De entre estos, el gen ACAN mapeado en 15q26.1, produce el Aggrecan, que es una proteína de 250 kDa a la cual se encuentran unidos unas 100 cadenas de condroitín sulfato y unas 30 de keratán sulfato. Tiene una función predominantemente estructural, ya que constituye el principal componente de la matriz del cartílago de los vertebrados, particularmente en el cartílago o placa de crecimiento (facilitando la formación ósea endocondral) y en el cartílago articular, proveyendo, gracias a sus características de gel hidratado, las condiciones necesarias para cumplir con la función de soporte de peso que realizan las articulaciones [6]. y S Dateki resume las características moleculares del Aggrecan y los hallazgos de por lo menos 25 mutaciones en el gen ACAN relacionadas con talla baja sindrómica y no sindrómica [7], y que agrupadas dan lugar a un conjunto de condiciones conocidas como Aggrecanopatías, y que incluyen principalmente la Displasia Espondiloepifisaria tipo Kimberley (SEDK) (Spondylo Epiphyseal Dysplasia Kimberley type), la Osteocondritis Disecante Familiar, la Macrocefalia con Displasia Epifisaria Múltiple y facies distintiva, Displasia Espondiloepimetafisaria tipo aggrecano y varios fenotipos idiopáticos de baja estatura [8].
La talla de un individuo es un rasgo cuantitativo y multifactorial [9,10], donde hay un componente genético relacionado a la talla de los padres que determinará un resultado equivalente al promedio de la talla de los progenitores, pero su estudio es complejo e implica necesariamente algoritmos diagnósticos en los que hay que tomar en cuenta muchos factores aparte de los genéticos que incluyen los nutricionales, endocrinológicos y ambientales entre otros [11].
Entre otros aspectos, estas características clínicas relevantes a considerar incluyen la simetría y proporción de los segmentos corporales del individuo, y cuando la desproporción de estos segmentos corporales puede ser evidenciada acompañando a una talla baja, es bastante probable que estemos frente a un posible diagnóstico de una displasia esquelética, las cuales tienen diferentes grados de severidad en cuanto a sus manifestaciones y morbimortalidad. La mayoría de las displasias esqueléticas son de origen genético y, por lo tanto, junto a la prolija evaluación clínica, incluyendo datos radiográficos y otros que resulten de importancia, el estudio genético molecular se convierte en una necesidad para llegar a un diagnóstico definitivo [11].
La Displasia Espondiloepifisiaria tipo Kimberley (SEDK), asociado al gen candidato por ligamiento genético por primera vez en 2002 en una familia de Sudáfrica, es una condición autosómica dominante caracterizada por baja estatura proporcionada, tronco y extremidades cortas y osteoartritis de inicio temprano [12]. y su nombre está asociado a que afecta principalmente las epífisis de los huesos largos y las vértebras. Otras manifestaciones incluyen hábito corporal rechoncho; a nivel vertebral los pacientes pueden presentar osteofitos anteriores, irregularidad de la placa vertebral terminal, cuerpos vertebrales escleróticos y platispondilia; a nivel pélvico, epífisis de la cabeza femoral aplanadas y a nivel de las extremidades osteoartropatía progresiva de inicio temprano, genu varo o genu valgo [13].
En nuestro caso, se detectó una variante probablemente patogénica (variante clase 2) en el gen ACAN no descrita previamente, que origina un codón de parada prematuro, el cual resulta en una degradación mediada sin sentido (Nonsense-mediated-degradation or NMD) del mARN del alelo mutante y, por lo tanto, se presume una haploinsuficiencia para el agrecano [14]. Las manifestaciones clínicas de nuestro paciente, aparte de la talla baja de - 5 DS, principal causa de consulta y preocupación, son relativamente leves, siendo lo más llamativo una frente ligeramente prominente y las extremidades inferiores ligeramente en varo (Figura 2).
El padre del probando, en el cual se detectó la misma mutación, tiene una talla de 1.61 m y dentro de nuestra población general pudiese pasar por una persona con una talla promedio un poco baja, pero no excesivamente llamativa, dentro del espectro posible de la población. Él único dato que a priori, nos pudiese haber hecho pensar en un caso de una displasia esquelética familiar, tampoco fue excesivamente grave y tenía que ver con los antecedentes de una cirugía de hernia de disco a los 43 años, y que pudiese estar relacionado con el estilo de vida de la persona u otros factores no genéticos.
En cuanto al tratamiento para SEDK, dado que estos pacientes presentan una edad ósea avanzada y un cese de crecimiento prematuro, la combinación del bloqueo de la pubertad por medio de Hormona liberadora de Gonadotropina (GnRH) y el uso oportuno de hormona de crecimiento (GH), ha resultado ser beneficiosa para los pacientes con esta condición, resultando en ganancias en su talla final [7].
La Osteoartritis (OA) es una de las principales causas de discapacidad y fuente de costo social en los adultos mayores. Es un trastorno degenerativo de las articulaciones sinoviales con una prevalencia estimada del 10 al 15 % de los adultos mayores de 60 años. Se considera que la degeneración del aggrecano está involucrada en esta condición tan común como parte central de su fisiopatología [15] [16]. Aunque su comprensión ha mejorado en los últimos años, todavía faltan por comprender por completo los mecanismos involucrados y por qué en los pacientes con SEDK la OA se presenta de forma prematura. Por ello los pacientes con esta condición y otras con estas variantes genéticas son objeto de estudio para aclarar parte de estos mecanismos y poder encontrar soluciones terapéuticas innovadoras, probablemente por parte de la medicina regenerativa con células madre [17].
CONCLUSIONES
Las pruebas de genética molecular se convierten en un aliado indispensable para completar algoritmos diagnósticos en casos de talla baja que resulten inexplicables luego de la determinación de factores endocrinológicos, nutricionales u otras causas no genéticas, ya que pueden revelar la presencia de variantes patogénicas en genes relacionados, permitiendo así el diagnóstico de displasias esqueléticas. Este reporte de caso nos muestra que existe un espectro de variabilidad en la talla de los individuos, que, a primera vista, no sugiere un diagnóstico de este tipo, pero que puede resultar explicativa y estar subyacente a una talla baja promedio acompañada de una gama de problemas ortopédicos, con un espectro de manifestaciones variable observados en algunos pacientes.
También este caso nos da un ejemplo más de la importancia de orientar el cambio de pensamiento en la práctica médica cotidiana. Esta debe encaminarse hacia el uso rutinario del diagnóstico molecular para aclarar el complejo panorama clínico. En esta ocasión, debido a variantes en una sola molécula, se pueden originar al menos cuatro entidades clínicas distintas y aparentemente no relacionadas, que forman parte de las Agrecanopatías. De igual manera en la práctica médica rutinaria, nos enfrentamos constantemente a entidades aparentemente distintas pero que tienen a una sola molécula como denominador común, como ya se ha demostrado en otros muchos casos.
El diagnóstico de SEDK, basado en alteraciones en el Aggrecan, proporciona una fuente de conocimiento para entender por qué en estos casos se presenta una degeneración acelerada del cartílago y encontrar así métodos terapéuticos más eficientes para tratar una condición con una incidencia tan alta como lo es la Osteoartritis (OA).
Agradecimientos
Gracias al Dr. Enrique A. Jiménez MD, FACC, FSCAI, Cardiólogo Intervencionista en Overton Brooks VA Medical Center, Shreveport, Louisiana; Director Asociado del Programa de Residencia de Cardiología en la Universidad Estatal de Luisiana en Shreveport, Luisiana, por sus contribuciones.
Gracias al Dr. Ernest Jermin, MD, Director Médico del Behavioral Health Center Florida en el Cleveland Clinic Indian River Hospital, por sus contribuciones.
Citas
[1] Couchman JR, Pataki CA. An introduction to proteoglycans and their localization. J Histochem Cytochem. 2012 Dec;60(12):885-97. doi: 10.1369/0022155412464638. Epub 2012 Sep 26.
[2] Lander AD, Selleck SB. The elusive functions of proteoglycans: in vivo veritas. J Cell Biol. 2000 Jan 24;148(2):227-32. doi: 10.1083/jcb.148.2.227.
[3] Xie C, Schaefer L, Iozzo RV. Global impact of proteoglycan science on human diseases. iScience 26, 108095, 2023, Nov; 1-19
[4] Iozzo RV, Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015 Mar; 42:11-55. doi: 10.1016/j.matbio.2015.02.003.
[5] Wight TN, Merrilees MJ. Proteoglycans in atherosclerosis and restenosis: key roles for versican. Circ Res. 2004 May 14;94(9):1158-67. doi: 10.1161/01.RES.0000126921.29919.51.
[6] Roughley PJ, Mort JS. The role of aggrecan in normal and osteoarthritic cartilage. J Exp Orthop 2014;1: 8.
[7] Dateki S. ACAN mutations as a cause of familial short stature. Clin Pediatr Endocrinol. 2017;26(3):119-125. doi: 10.1297/cpe.26.119.
[8] Gibson BG, Briggs MD. The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis. 2016 Jun 28;11(1):86. doi: 10.1186/s13023-016-0459-2.
[9] Lello L, Avery SG, Tellier L, Vazquez AI, de Los Campos G, Hsu SDH. Accurate Genomic Prediction of Human Height. Genetics. 2018 Oct;210(2):477-497. doi: 10.1534/genetics.118.301267. Epub 2018 Aug 27. Erratum in: Genetics. 2020 Jan;214(1):231
[10] Lobo, I. Multifactorial inheritance and genetic disease. Nature Education 2008. 1(1):5
[11] Quintos JB, Guo MH, Dauber A. Idiopathic short stature due to novel heterozygous mutation of the aggrecan gene. J Pediatr Endocrinol Metab. 2015 Jul;28(7-8):927-32. doi: 10.1515/jpem-2014-0450.
[12] Anderson IJ, Tsipouras P, Scher C, Ramesar RS, Martell RW, Beighton P. Spondyloepiphyseal dysplasia, mild autosomal dominant type is not due to primary defects of type II collagen. Am J Med Genet 1990;37: 272–6. doi: 10.1002/ajmg.1320370223
[13] Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 608361: 12/24/2007. World Wide Web URL: https://www.omim.org/entry/608361?search=kimberley%20type&highlight=kimberley%2Ctype
[14] Schweingruber C, Rufener SC, Zund D, Yamashita A, Muhlemann O. Nonsense-mediated mRNA decay - mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta. 2013;1829(6–7):612–623. doi: 10.1016/j.bbagrm.2013.02.005
[15] Grässel S, Aszodi A. Osteoarthritis and Cartilage Regeneration: Focus on Pathophysiology and Molecular Mechanisms. Int J Mol Sci. 2019 Dec 6;20(24):6156. doi: 10.3390/ijms20246156.
[16] Alberton P, Dugonitsch HC, Hartmann B, Li P, Farkas Z, Saller MM, Clausen-Schaumann H, Aszodi A. Aggrecan Hypomorphism Compromises Articular Cartilage Biomechanical Properties and Is Associated with Increased Incidence of Spontaneous Osteoarthritis. Int J Mol Sci. 2019 Feb 26;20(5):1008. doi: 10.3390/ijms20051008.
[17] G-I. Im Current status of regenerative medicine in osteoarthritis. Bone Joint Res 2021;10(2):134–136. doi: 10.1302/2046-3758.102.BJR2020-0517.R1
