INTRODUCCIÓN
Las anomalías craneofaciales son un grupo de malformaciones congénitas raras, con una prevalencia de 88.4 por cada 100,000 nacimientos y diversas etiologías. Un síndrome diagnosticado comúnmente con características craneofaciales es la microsomía craneofacial.
La microsomía craneofacial (MCF; OMIM#164210) es la forma actual de designación para una condición que ha recibido diferentes nombres y caracterizaciones a lo largo de los años. Ya sea llamada MCF, síndrome de Goldenhar [2], espectro oculo-auriculo-vertebral [3] o microsomía hemifaceal [4], se sabe que esta condición consiste en una malformación congénita asimétrica de los arcos branquiales primero y segundo, posiblemente debido a una lesión vascular o migración alterada de las células de la cresta neural [5].
Así, todas las estructuras derivadas de estos arcos pueden verse afectadas, resultando en un amplio espectro de características clínicas, que abarcan oídos, ojos, músculos, huesos faciales y vértebras cervicales [6].
El amplio espectro fenotípico impulsa a los médicos a considerar otros síndromes con anomalías craneofaciales como el síndrome de Treacher Collins, el síndrome branquio-oto-renales, la disostosis maxilofacial y el síndrome del ojo de gato [7].
El Síndrome del Ojo de Gato (CES; OMIM#115470) ocurre debido a una tetrasomía parcial del cromosoma 22, como resultado de un marcador supernumerario dicéntrico con satélites en ambos extremos, inv dup(22) (pter->q11.2::q11.2->pter) [8,9]. Este brazo cromosómico tiene 8 regiones de repeticiones de baja copia (LCR) llamadas LCR22A a LCR22H, y su homología predispone a la formación de reorganizaciones [10]. El CES se caracteriza clínicamente por un fenotipo muy variable con la presencia de múltiples malformaciones, que afectan principalmente a los ojos, oídos, regiones anorrectales y urogenitales [8]. La región genómica relacionada con esta comorbilidad se mapea entre el centrómero y el LCR22A y se denomina Región Crítica del Síndrome del Ojo de Gato (CESCR) [11]. Existen dos tipos de CES: el tipo I se caracteriza por un punto de ruptura en o antes del LCR22A, y el tipo II incluye la región crítica del síndrome de DiGeorge (DGS) [12].
Aquí informamos sobre un paciente clínicamente mal diagnosticado con CFM, siguiendo la lista de verificación CARE [13], que tiene pruebas de laboratorio compatibles con el tipo II de CES. Para discutir nuestros hallazgos, realizamos una revisión de la literatura. Como los cromosomas del marcador de ella divergieron ligeramente con los encontrados en publicaciones recientes, lo ampliamos a 20 años de casos e informes de series de pacientes diagnosticados con CES, que destacan la rareza de nuestro caso. También discutimos y propusimos un mecanismo para la formación de esta alteración cromosómica compleja. Por último, informamos brevemente sobre el fenotipo de casos previamente no publicados en Brasil y examinamos las preocupaciones epidemiológicas y clínicas.
INFORME DE CASO
La paciente es una niña, el primer hijo de una pareja joven, sana y no consanguínea de la región sur de Brasil. El embarazo fue normal y no hubo casos de dismorfismos faciales en la historia familiar (pedigrí de tres generaciones). Nació a las 35 semanas y 1 día, con un bajo peso al nacer (1920 gramos; por debajo del percentil 10). En la sala de partos, el pediatra informó sobre anomalías en las orejas. No se reportaron anomalías abdominales en el examen físico y la ecografía.
Una tomografía computarizada a 1 mes reveló hipoplasia mandibular derecha y anomalías en el conducto auditivo, incluyendo posible fusión del martillo y yunque, y dislocación de estos huesos hacia la membrana timpánica lateral superior. Las pruebas de respuesta auditiva evocado del tronco encefálico también mostraron disfunción en el oído derecho.
A los 2 años, el paciente desarrolló infecciones urinarias recurrentes y tuvo convulsiones febril en tres ocasiones. La gammagrafía renal con Tc-99m ácido dimercaptosuccínico fue normal, y no se observaron anomalías anatómicas en la ecografía; se prescribió profilaxis antibiótica. Un EKG mostró un intervalo PR acortado (0,09s; HP: 0005165), pero sin repercusiones clínicas ni anormalidades en la ecografía.
A los 3 años, la paciente fue referida al servicio ambulatorio de genética médica y evaluada por un experimentado dismorfólogo. Se detectó un retraso en el desarrollo neuromotor. Tenía un peso bajo (por debajo del percentil 5; HP: 0004325), cabello rizado (HP: 0002212), microtia derecha (HP: 0008551), microftalmía derecha (HP: 0000568), pliegue epicántico derecho (HP: 0000286), puente nasal ancho (HP: 0000431), paladar alto (HP: 0000218), etiqueta de piel preauricular derecha (HP: 0000384) y fosita (HP: 0100277), y hendidura sacra (HP: 0000960). No se notaron dermoides epibulbares, fisura oral ni ninguna otra anomalía física, y la evaluación oftalmológica fue sin incidentes. Dado que la paciente presentaba dismorfismos faciales asimétricos, se propuso la hipótesis de CFM.
Se realizó un cariotipo con bandas G, identificando 47,XX,+mar[25]. La hibridación genética comparativa basada en matriz (array-CGH - CytoScan™ 750K Suite - Thermo Fisher Scientific, Catálogo #901859) confirmó que el marcador extranumerario derivaba de un cromosoma 22 y delimitaba un punto de quiebre LCR22B (Figura 1). Así, se confirmó el tipo II CES asimétrico, y el resultado citogenético completo se definió como 47,XX,+mar[25]. arr[GRCh37] 22q11.1q11.21 (16888899_18644240 x4, 18649189_20716903x3,20723685x2).
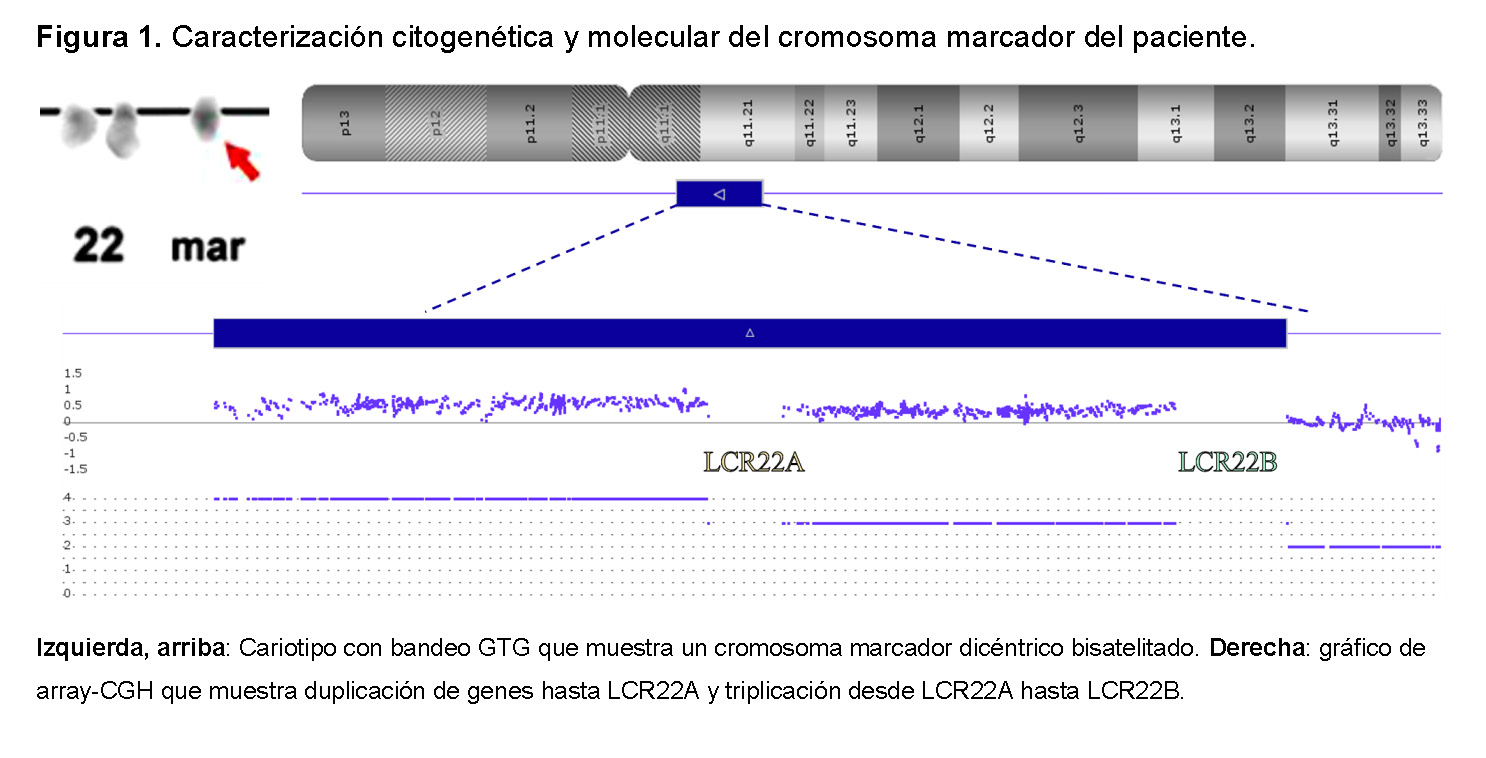
A los 10 años, al paciente también se le diagnosticó trastorno por déficit de atención e hiperactividad.
DISCUSIÓN
Nuestra hipótesis clínica original era CFM, pero los hallazgos citogenéticos y moleculares establecieron el diagnóstico de CES. En un intento por determinar el diagnóstico del paciente, nos encontramos con la similitud de la manifestación clínica de ambos síndromes, que involucra principalmente fositas preauriculares, microtia y dismorfismos faciales asimétricos.
La tríada clásica del CES son anomalías preauriculares, malformación anal y coloboma de iris. Por lo tanto, nuestro paciente tiene solo una característica clásica (anomalía preauricular). En una cohorte con pacientes que tienen ganancia en la región LCR22A, la mayoría de los individuos tienen solo una o dos características descritas en la tríada clásica (75%) [12].
Por lo tanto, puede que no sea útil para identificar la mayoría de los casos de CES.
Asimismo, considerando los criterios diagnósticos de CFM (los criterios FACIAL y ICHOM descritos por Renkema et al.) [14], nuestro paciente podría ser diagnosticado erróneamente con CFM si no se identifica la ganancia citogenética/la identificación de otros síndromes, establecidos como criterios de exclusión.
Como consecuencia de esta superposición clínica, no es raro encontrar individuos con CES y diagnóstico clínico de CFM. En la literatura, podemos encontrar algunos informes, incluido un estudio reciente realizado por Spineli-Silva et al. [6], que exploró la relación entre los dos fenotipos basándose en informes de casos de tres pacientes. Así que, incluso si el CFM es considerado por algunos autores [14], el CES debe ser considerado como un diagnóstico diferencial.
Este solapamiento fenotípico y la necesidad de análisis citogenéticos pueden ser una barrera diagnóstica significativa en un país subdesarrollado y emergente como Brasil. Según la Red Brasileña de Enfermedades Raras (BRDN; una base de datos poblacional de enfermedades raras), 19 pacientes con posible CFM son atendidos por genetistas clínicos, y 20 pacientes han sido confirmados con esta condición. Ninguno de ellos reportó pruebas citogenéticas y/o moleculares, a pesar de que los criterios FACIAL e ICHOM sugieren que se deben descartar otros síndromes. Además, solo se informa que cinco pacientes más tienen un diagnóstico de CES confirmado.
Debe destacarse que, aunque las instituciones que componen el BRDN brindan atención a la mayoría de la población brasileña, proporcionan, en promedio, 84 pruebas de microarreglo y 47 pruebas de hibridación in situ por fluorescencia (FISH) por mes [15]. Así, la baja capacidad puede explicar la investigación incompleta de la mayoría de los casos.
Entre los pacientes diagnosticados con CES, solo uno tenía la tríada clásica, uno tenía dos de los signos (anomalía anal más coloboma), y los tres restantes no tenían ninguno. Las manifestaciones clínicas de estos pacientes se pueden ver en la Tabla 1; todos los casos fueron de novo (no recidiva familiar en un pedigrí de tres generaciones).

Aunque no tenemos suficiente información para determinar el tipo de CES con el que se había diagnosticado a estos pacientes, podemos inferir a partir del amplio espectro fenotípico que la evaluación clínica es insuficiente para establecer el diagnóstico, como se ha informado anteriormente en la literatura [12]. Por lo tanto, los hallazgos de laboratorio son esenciales para confirmar el diagnóstico.
Dado que los dismorfismos faciales se pueden observar desde el nacimiento y pocos casos se identifican prenatales (0.6% [1]), creemos que los pediatras y neonatólogos deberían ser capaces de identificar posibles casos de CFM, CES y otros síndromes con anomalías craneofaciales, y referir adecuadamente a estos pacientes a genetistas clínicos, quienes podrán proporcionar asesoramiento genético.
Este desafío está relacionado con la Política Nacional Brasileña para la Atención Integral en Genética Clínica. A pesar de que se ha establecido durante más de una década, el sistema de salud pública aún carece de vías clínicas para investigar y manejar enfermedades raras. Además, esta ordenanza legislativa no fue seguida por una ejecutiva; por lo tanto, no existe un plan financiero nacional, consolidado y a largo plazo.
Estos problemas políticos y administrativos, además del fenotipo variable discutido anteriormente, pueden explicar la larga odisea del paciente de 16 años.
Revisión de literatura
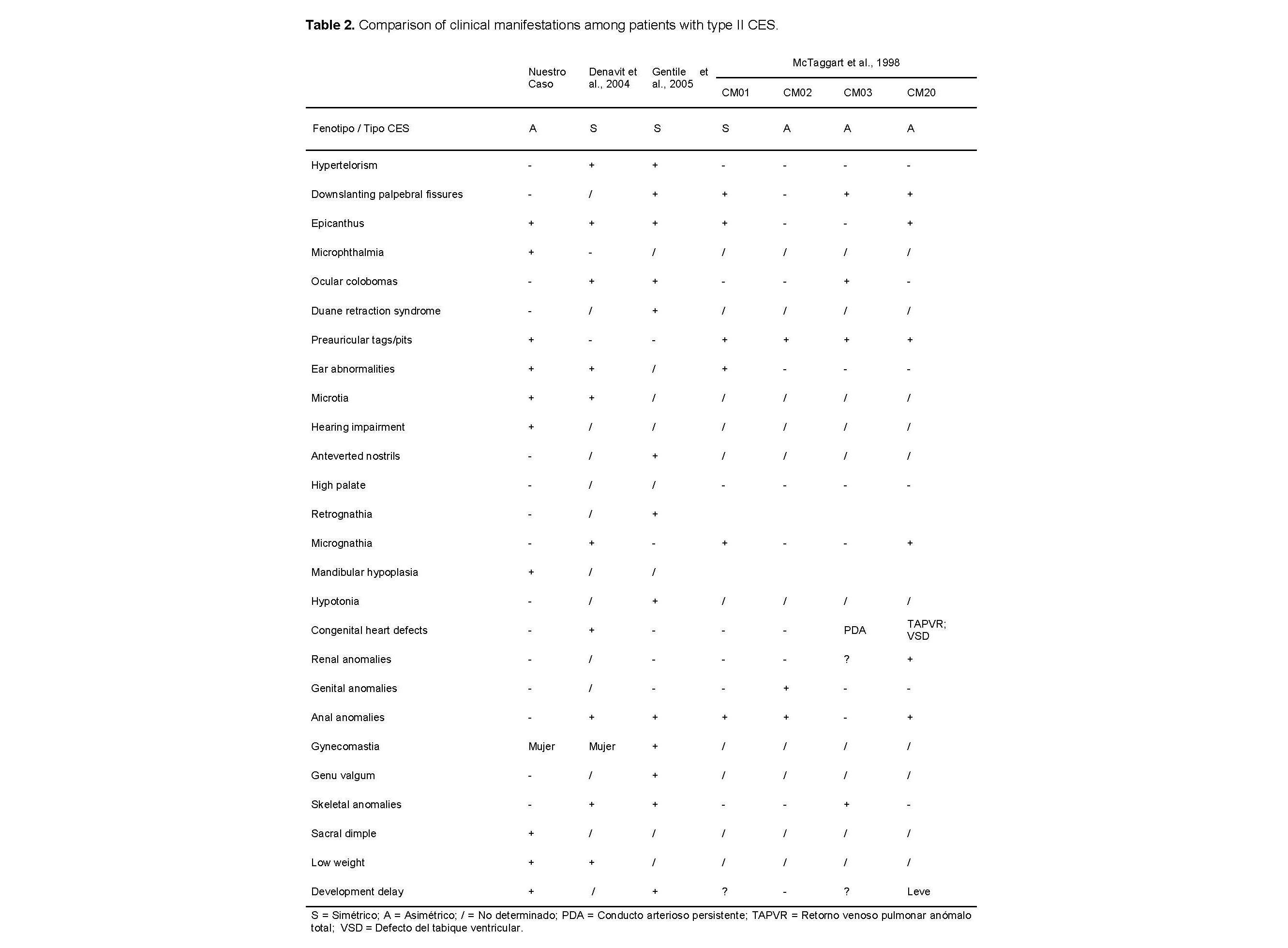
Nuestro caso tiene una duplicación que va desde pter hasta LCR22B. Cuando se compara con investigaciones recientes sobre CES, todos los casos reportados [6] tienen la porción duplicada que va desde pter hasta LCR22A (que incluye los genes hasta USP18). A pesar de esta diferencia en los loci (que involucra 30 genes), no se observó una variación fenotípica significativa.
Así, decidimos ampliar nuestra búsqueda de literatura, buscando pacientes con CES con caracterización molecular. En los últimos 20 años, se han reportado un total de 53 casos [17–47], y solo dos se encontraron con un cromosoma marcador más grande; sin embargo, a diferencia de nuestro informe, ambos eran simétricos.
El primero muestra alteraciones craniofaciales severas, incluyendo la ausencia de ambas orejas [47]. El segundo [45] manifiesta el síndrome de retracción de Duane, un hallazgo reportado en seis otros casos de CES, pero en el tipo I [43,44,32,29,31].
Las manifestaciones clínicas de estos pacientes se comparan con nuestro paciente en la Tabla 2. También comparamos los hallazgos morfológicos con los de la primera serie de casos [48], añadiendo cuatro casos más de CES tipo II a nuestro conjunto.
Síndrome del Ojo de Gato Crítico Región del Cromosoma
La región cromosómica del síndrome del ojo de gato (CESCR) está mapeada entre el centrómero del cromosoma 22 y LCR22A [coordenadas genómicas (GRCh38/hg38): 22:17,359,949–17,558,151]. Esto explica las manifestaciones clínicas de aquellos pacientes reportados con el punto de ruptura en o antes de esta región (Tabla Suplementaria 1). Hasta la fecha, se han identificado 14 genes en el CESCR [23], y los individuos con duplicaciones o triplicaciones intracromosómicas de esta región han mostrado fenotipos como el CES, así como cromosomas en anillo [49–53,39], lo que indica que la trisomía parcial es suficiente para causar las características mayores y menores del CES, independientemente de la estructura del marcador cromosómico extranumérico [54]. Aún así, todavía hay algunos casos de CES clínicamente definidos o convincentes cuya etiología genética permanece por elucidarse [12].
A diferencia de latrisomía, la haploinsuficiencia de estas regiones parece ser inofensiva [55], ya que la trisomía de los genes después de LCR22A no parece producir un fenotipo más severo que el asociado con el tipo I de CES. Por lo tanto, la determinación del tipo de ruptura de CES no tendría valor pronóstico [54]. Pero, ¿es realmente así?
Aunque se ha sugerido que la inclusión de genes después de LCR22A no contribuye al fenotipo de CES, se han descrito duplicaciones en esta región en tres pacientes con un resultado clínico similar: extrofia de vejiga. El primer caso, un niño no sindrómico con extrofia de vejiga clásica, tenía una duplicación que abarcaba desde los genes CLTCL1 hasta IZTR1, lo que indica un rearrangement LCR22A–LCR22D [56]. Los otros dos casos eran pacientes no relacionados con el mismo resultado más deterioro auditivo, con regiones duplicadas descritas como arr.[NCBI36/hg18]17313883_19854524 en el caso 1 y arr.[NCBI36/hg18]17313883_20006849 en el caso 2, ambos también rearrangements que involucraban LCR22A–LCR22D [57].
El mecanismo propuesto para la extrofia de vejiga implica la sobreexpresión de DGCR8 interfiriendo con la regulación de p63 a través del procesamiento de microRNA [56]. Sin embargo, nuestro paciente tiene DGCR8 duplicado y no muestra ningún signo de extrofia de vejiga, lo que indica que la contribución genómica a este resultado probablemente se encuentra más allá de LCR22B. Considerando otras anomalías urinarias, alrededor del 8% de los pacientes con CES pueden tener reflujo vesicoureteral [12], pero aún no hay una explicación satisfactoria para esta asociación.
Intrigantemente, la variación en el número de copias (CNV) del área de DiGeorge puede tener algún impacto clínico después de todo. Esta condición es conocida como el síndrome de duplicación 22q11.2 (OMIM #608363), y sus características fenotípicas se superponen con las del síndrome de DiGeorge (deleción 22q11.2). El principal gen relacionado con esta condición es TBX1, un miembro de la familia de genes T-box, cuya sobre- o subexpresión se ha implicado en el espectro completo de malformaciones del síndrome de DiGeorge [56]. Aún así, TBX1, junto con CLTCL1, GSC2, HIRA y MAPK1, forma un grupo de genes sugeridos como candidatos para estar involucrados en la naturaleza asimétrica de la etiología de la microsomía craneofacial [58]. Todos estos genes están ubicados entre LCR22A y LCR22B. En consecuencia, están duplicados en nuestro paciente, pero no están duplicados en los casos más comunes de CES, a saber, el CES tipo I.
Otro estudio que investiga microduplicaciones en el cromosoma 22q11.2 analizó 31 casos, incluyendo tres con duplicaciones de LCR22A a LCR22B, cuatro de LCR22B a LCR22D, cinco de LCR22C a LCR22D, dieciséis de LCR22A a LCR22D, y tres en regiones LCR distales (una LCR22E–LCR22F y dos LCR22E–LCR22H). Curiosamente, los individuos con duplicaciones que involucran LCR22A–B y LCR22B–D mostraron un desarrollo normal en el examen físico; sin embargo, se documentaron anomalías renales en tres fetos, todos involucrando LCR22D, lo que sugiere que las duplicaciones en esta región podrían desempeñar un papel clave en los fenotipos renales. Además, se observaron fenotipos raros como anomalías esqueléticas, anomalías faciales e hipoplasia tímica, indicando que las alteraciones en la dosis génica en la región 22q11.2 son determinantes críticas de las manifestaciones clínicas. Estas variaciones en los resultados clínicos pueden reflejar la influencia de la configuración genómica o los efectos posicionales de los genes sobre el fenotipo.
Mecanismos de duplicación
Los mecanismos más aceptados implicados en los reordenamientos de CES se basan en eventos de recombinación homóloga (HR) y recombinación homóloga no alélica (NAHR) entre los LCR22 durante la meiosis, así como en duplicaciones y deleciones dentro de la región 22q11.2 [60,61,20]. La razón de tal multitud de reordenamientos radica en la complicada orientación invertida y directa de las secuencias en los LCR22 [60]. Sin embargo, la formación de un marcador bisatelitado y estructuras de duplicación intersticial es ligeramente diferente. Los marcadores simétricos tipo I y tipo II de CES resultan de un emparejamiento correcto de regiones de cromosomas homólogos o de cromátidas hermanas seguido de una resolución errónea, mientras que las duplicaciones asimétricas tipo II de CES y las duplicaciones intersticiales resultan de NAHR, lo que significa que el emparejamiento ocurre entre diferentes LCRs (Figura 2A). Una forma de verlo es que las duplicaciones intersticiales son recombinaciones asimétricas tipo II seguidas de una resolución correcta. En esos casos, generalmente solo uno de los dos centrómeros es funcional y es tirado al azar por la fibra del huso [20].
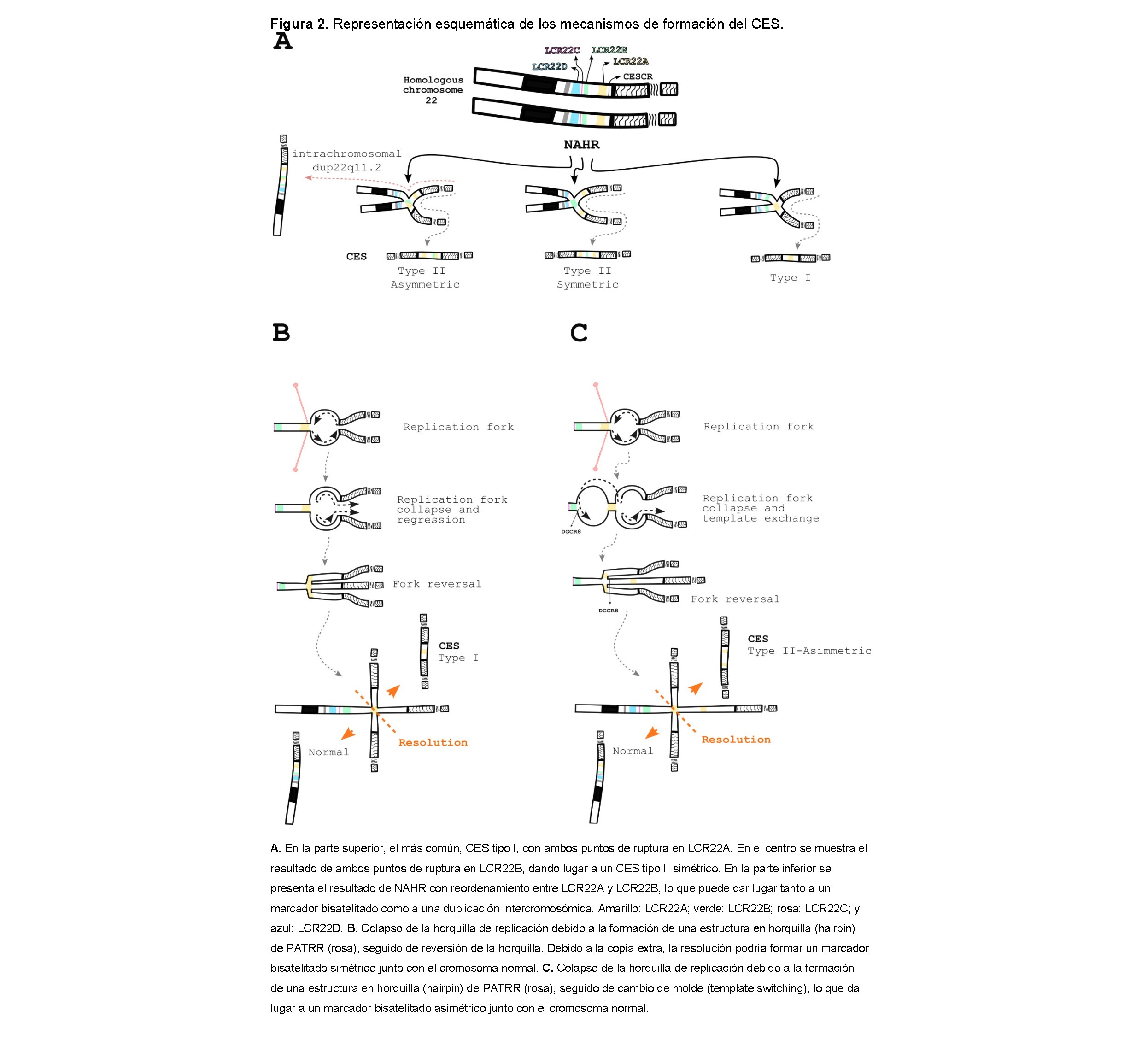
Teóricamente, la reorganización podría ocurrir entre cualquier LCR22, pero en nuestra revisión, no nos hemos encontrado con ningún caso más allá de LCR22B en un marcador cromosómico bisatélite. La duplicación intercromosómica, por otro lado, se ha descrito con genes duplicados, ya sea en orientación directa o invertida, a lo largo del brazo largo del cromosoma 22.
Un elemento estructural de ADN en particular, un repeticiones palindrómicas ricas en AT (PATRR), manifiesta polimorfismos de tamaño y está presente en LCR22A, LCR22B y LCR22D, rodeado por un subtipo de satélite humano I (HSAT I) y un elemento AluSc, formando una estructura tripartita de 2.4 kb. La simetría y longitud de los PATRR influyen en gran medida en la frecuencia de translocaciones de novo, debido a su tendencia a formar una conformación de ADN cruciforme in vivo, lo que puede inducir rupturas y, en consecuencia, uniones ilegítimas. Se ha sugerido que estos PATRR están involucrados en translocaciones recíprocas que incluyen las reordenaciones recurrentes t(11;22)(q23;q11.2), t(17;22) (q11.2;q11.2) y t(8;22)(24.1;q11.2), así como eventos no recurrentes como t(4;22)(q35.1;q11.2) y t(1;22)(p21.1;q11.2).
LCR22A y LCR22D son las regiones LCR más grandes, lo que puede explicar su mayor frecuencia de reordenamiento. Como se puede observar, el tipo I CES es de lejos el más común. Sin embargo, la mayoría de las translocaciones mencionadas anteriormente involucran LCR22B, lo que hace que esta hipótesis sea menos probable.
Un hecho intrigante es que HR o NAHR entre cromosomas homólogos no daría lugar a un cromosoma 22 normal capaz de segregar. Así, en este escenario, no sería posible obtener un cariotipo con un marcador bisatelítico extranumerario. Por otro lado, los reordenamientos entre cromátidas hermanas permitirían esto. Pero, ¿hay otra opción?
Los pasadores de cabello que se forman en los extremos de ADN de cadena simple expuestos por resección pueden inhibir la reparación eficiente del ADN [65]. Otra explicación que viene a la mente no involucra una ruptura, sino un colapso de la horquilla de replicación. En este escenario, una horquilla retrocedida promueve una copia extra y forma una unión de Holliday, en la cual el proceso de reparación genera no solo el marcador bisatélite, sino también un cromosoma 22 normal (Figura 2B–C). Según los mecanismos basados en la replicación, esta yuxtaposición de secuencias de ADN no contiguas puede variar desde unos pocos cientos de pares de bases hasta megabases y puede ser impulsada por tan solo dos a seis nucleótidos de homología [66].
Este mecanismo podría ser una explicación plausible para aquellos casos de CES en los que el punto de ruptura ocurre antes de LCR22A, así como para los CES simétricos de tipo I y II, ya que el colapso podría ser causado por una configuración de ADN no B de PATRR (Figura 2B). También podrían surgir CES asimétricos, pero con un paso adicional de intercambio de plantilla hacia una horquilla de replicación proximal, que en este caso estaría situada cerca de LCR22B (Figura 2C).
Independientemente, las diferencias en el apareamiento de cromosomas pueden llevar a diferencias en el contenido genético. En casos de CES simétrico, todos los genes en el cromosoma marcador serán cuadruplicados, con un conjunto en una configuración invertida. Mientras tanto, el CES tipo II asimétrico tendrá solo los genes hasta LCR22A cuadruplicados y los de LCR22A a LCR22B triplicados, también con un conjunto en una configuración invertida. Estas variaciones en el contenido genético y la orientación de los genes pueden llevar a diferentes combinaciones de expresión génica que podrían influir en las manifestaciones clínicas.
CONCLUSIÓN
Informamos un caso de CES y reflexionamos sobre los aspectos clínicos, epidemiológicos y moleculares de esta enfermedad. Dado que solo una minoría de los pacientes presenta la tríada clásica, se debe considerar el CES como un diagnóstico diferencial del CFM. Además, esta superposición clínica puede dificultar el diagnóstico en países subdesarrollados, donde las investigaciones de laboratorio son esenciales para confirmar el CES.
Respondiendo a nuestra pregunta principal, parece que, al menos en este caso, el tamaño realmente no importa. Esto sugiere que un aumento en la variación en el número de copias (CNV) que abarca todo el conjunto de genes entre LCR22A y LCR22B, como sugieren otros informes, no desempeña un papel importante en las manifestaciones clínicas del CES. Sin embargo, si estos genes supuestamente están implicados en el CFM y otras manifestaciones fenotípicas, ¿por qué su sobreexpresión no se traduce en hallazgos clínicos? Una posible explicación podría residir en la configuración genómica o el contexto posicional de los genes dentro del cromosoma.
La definición clásica de CES infiere la presencia de una estructura de marcador cromosómico, pero hallazgos más recientes sobre CNVs dentro del CESCR [49–53,39] sugieren que la estructura en sí misma puede no ser el factor determinante. A pesar de esto, queda una pregunta importante: ¿qué camino se debe seguir para comprender mejor la etiología de la enfermedad?
Contrario a lo que se ha propuesto anteriormente, nuestros hallazgos sugieren que el mecanismo más probable puede ser basado en la replicación, ya que tal mecanismo podría generar ambos cromosomas simultáneamente: un cromosoma 22 normal y un cromosoma marcador bisatelital. Por lo tanto, sería muy informativo investigar los puntos de quiebre y el contenido genético de los marcadores bisatelitales en detalle, como se demostró en nuestro caso utilizando una sonda TXNRD2. Al hacerlo, podría ser posible extrapolar estos hallazgos a otros trastornos causados por estructuras cromosómicas similares, como el síndrome inv dup(15), mejorando en última instancia la atención al paciente a través de correlaciones más precisas entre genotipo y fenotipo y mejores consejerías genéticas.
Declaración de reconocimiento
Nos gustaría agradecer a la paciente y a su familia. Este estudio fue apoyado por InRaras - Instituto Nacional de Enfermedades Raras.
Declaración de contribución del autor
RM: Conceptualización, Curación de datos, Escritura – borrador original; JPPT: Conceptualización, Curación de datos, Escritura – borrador original; MM: Curación de datos, Escritura – borrador original; LDW: Curación de datos, Escritura – borrador original; GA: Análisis formal, Metodología, Escritura – revisión y edición; PT: Validación, Escritura – revisión y edición; BMO: Supervisión, Escritura – revisión y edición; TMF: Supervisión, Escritura – revisión y edición; PRGZ: Supervisión, Administración del proyecto, Escritura – revisión y edición.