
INTRODUCCIÓN
La deleción 22q11.2, también conocida como síndrome de DiGeorge (22q11DS, OMIM #192430, #188400), es un trastorno multisistémico que puede llevar a una amplia gama de manifestaciones clínicas asociadas con cardiopatías congénitas, paladar hendido, endocrinopatías, convulsiones, inmunodeficiencias, discapacidad intelectual y características neuropsiquiátricas [1,2]. Esta deleción afecta aproximadamente a 1 de cada 2,148 nacimientos vivos, se estima en 1 de cada 992 embarazos no seleccionados y 1 de cada 1,497 abortos espontáneos, lo que la convierte en uno de los trastornos genéticos más comunes [3]. El origen de esta enfermedad es muy complejo, y para entenderlo mejor, abordamos aquí algunas preguntas cruciales.
¿Por qué ocurre el 22q11DS?
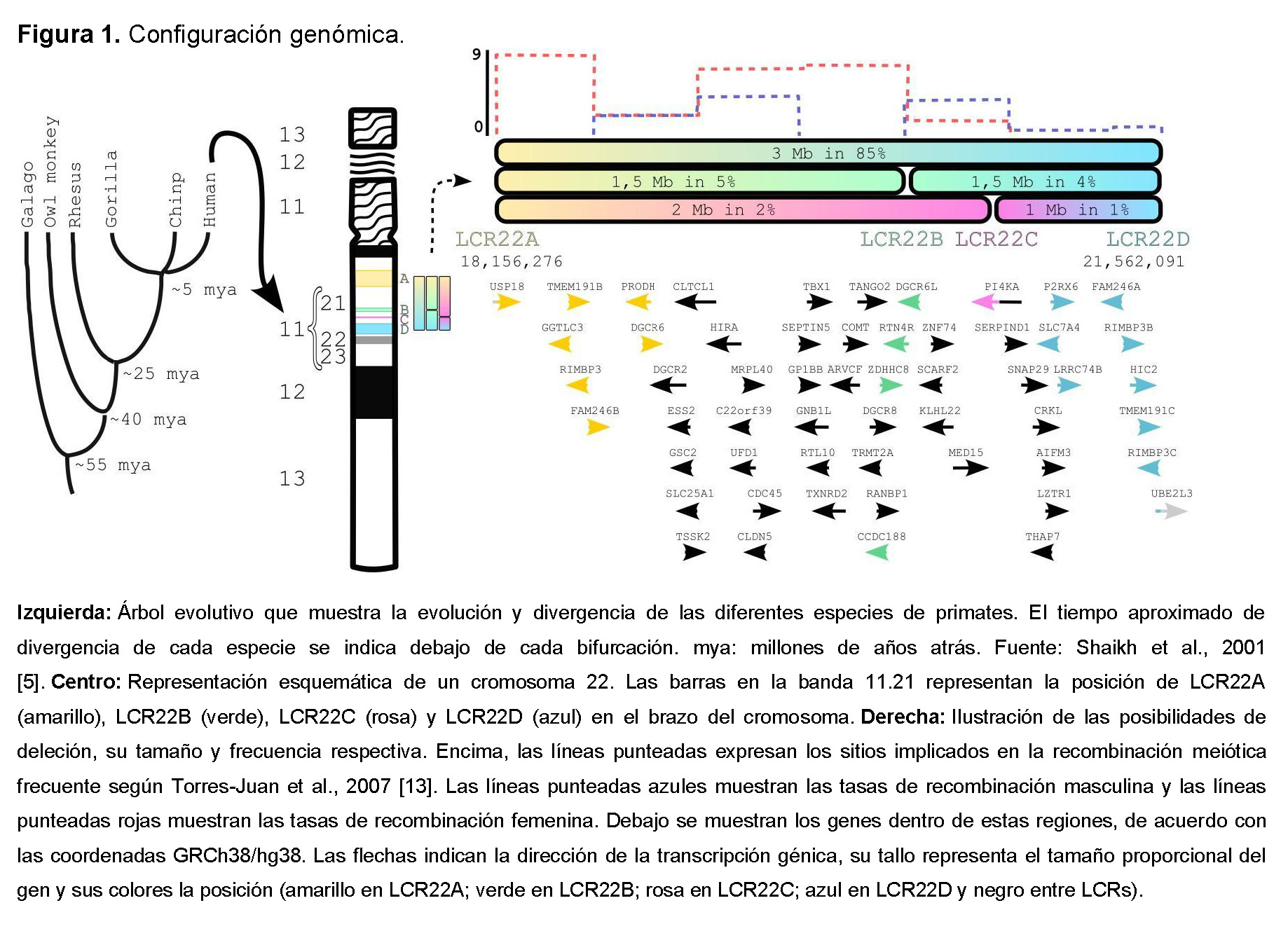
Hay ocho grupos de repeticiones de baja copia (LCR) distribuidos a lo largo del brazo largo del cromosoma 22. Se denominan LCR22A a LCR22H [4] y se originaron hace al menos 40 millones de años a través de reordenamientos mediados por Alu [5,6]. La presencia de estos LCRs hace que esta región sea altamente inestable, lo cual es consistente con la alta incidencia de esta condición.
La deleción típica involucra LCR22A y LCR22D (3 Mb) y ocurre en el 85% de los individuos. Cuando están involucrados LCR22F–LCR22B y/o LCR22C, las deleciones se definen como "anidadas". Alrededor del 5% de los individuos afectados portan la deleción LCR22A–LCR22B (1.5 Mb), 2% LCR22A–LCR22C (2 Mb), 4% LCR22B–LCR22D (1.5 Mb), y 1% LCR22C–LCR22D (1 Mb) (Figura 1). Las deleciones que involucran LCR22E a LCR22H son menos frecuentes, con fenotipos que difieren del 22q11.2DS [4,7]; por lo tanto, nos centraremos solo en las deleciones de LCR22A a LCR22D.
¿Cómo se genera?
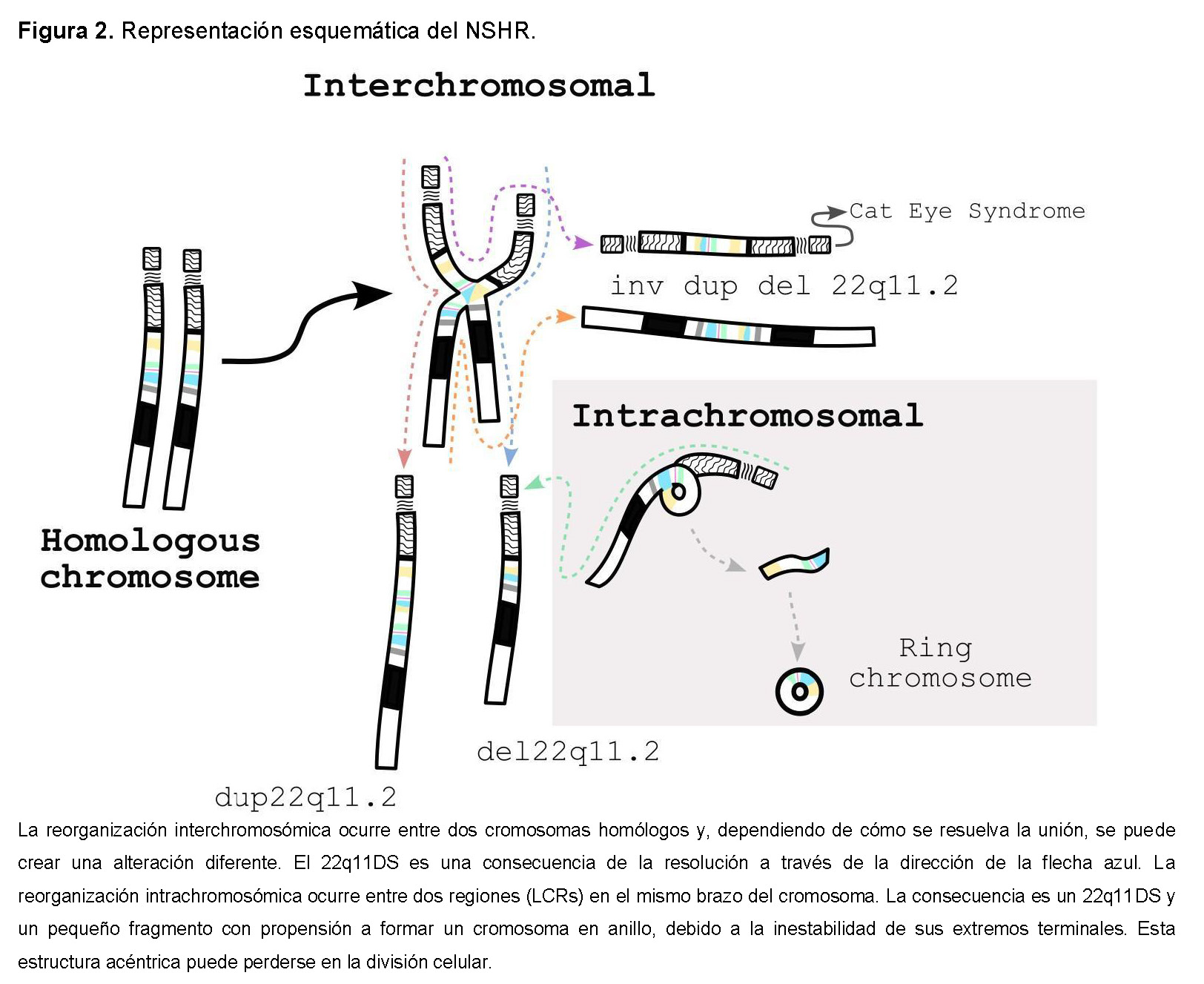
Se genera a través de la recombinación homóloga no alélica (NAHR) intercromosómica o intracromosómica. Considerando que estos LCRs comparten más del 96% de homología, la NAHR es el mecanismo más probable [4,7]. Cada combinación puede resultar en diferentes alteraciones cromosómicas, incluidas duplicaciones (síndrome de duplicación 22q11.2, OMIM #608363), inversiones, cromosomas marcadores bisatelitados supernumerarios (síndrome del ojo de gato, OMIM #115470) y cromosomas en anillo, además de deleciones (Figura 2).
¿Quién lo causa?
Puede ser heredado de padres portadores de la deleción o causado por errores en las divisiones de gametos o embriones (Figura 3). Aproximadamente el 90–95% de los casos son deleciones de novo, con ambos sexos y todos los grupos étnicos afectados, y una ligera predominancia de origen materno [8]. No hay asociación con la edad parental [2,4,7]. Incluso cuando ninguno de los padres tiene la deleción, todavía hay un riesgo de recurrencia (RR) ligeramente aumentado debido al mosaicismo o mosaicismo de línea germinal. Se sospecha mosaicismo de línea germinal cuando dos o más niños afectados nacen de padres aparentemente no afectados [9]. En estos casos, el RR es difícil de estimar debido a múltiples modos posibles de segregación [10].
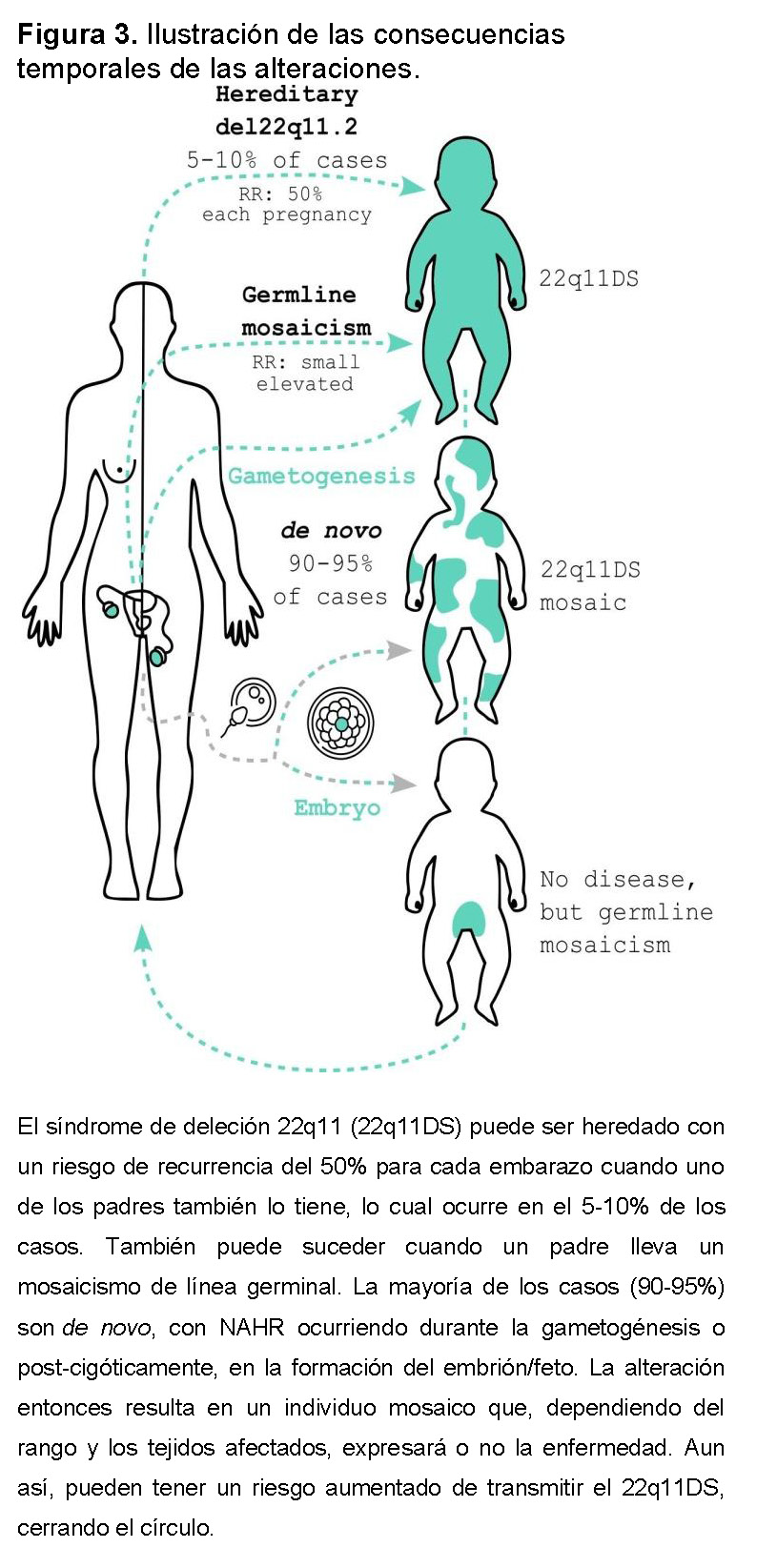
El 5–10% restante de los casos son heredados de un padre que puede no ser consciente de su condición genética. Una revisión completa de las características asociadas en la edad adulta se puede encontrar en Boot et al., 2023 [11]. Un portador de la deleción tiene un 50% de probabilidad de transmitirla en cada embarazo. Parece haber una fuerte presión selectiva negativa contra las deleciones 22q11.2 mediada por la severidad del fenotipo neuropsiquiátrico, pero no necesariamente por enfermedades cardíacas congénitas graves [12].
¿Cuándo se desarrolla?
La deleción surge durante la meiosis o la mitosis. Se ha asumido que la NAHR meiótica es el principal mecanismo en la formación de estas alteraciones cromosómicas. Curiosamente, la tasa de recombinación femenina en la región 22q11.2 es aproximadamente 1.6 a 1.7 veces mayor que en los hombres, lo que sugiere un sesgo de sexo en los procesos meióticos que puede explicar la predominancia de origen materno [8]. Un estudio previo respalda esta observación, mostrando que los LCR22 implicados en deleciones y duplicaciones son sitios de recombinación meiótica frecuente, principalmente durante la oogénesis (Figura 2, línea punteada) [13].
No hay evidencia de que la presencia de una alteración predisponga a otra. Sin embargo, se ha reportado una deleción anidada de novo (entre LCR22A y LCR22B) junto con una duplicación en el 70% de las células de una niña. Su padre, así como otros miembros de la familia, también portan una duplicación (entre LCR22B y LCR22D).
En este caso, el mecanismo propuesto fue mitótico en lugar de meiótico, involucrando un cruce desigual durante una división temprana después de la fertilización [14].
Así, desde otra perspectiva, la tasa de mutación de novo podría estar relacionada con la reparación ineficaz de lesiones espontáneas del ADN [9], ya que el ADN está sujeto a numerosos eventos dañinos diariamente como resultado del metabolismo celular normal, y lesiones adicionales pueden ser generadas por agentes exógenos [15]. Hasta la fecha, ningún estudio ha correlacionado el daño y la reparación del ADN con la ocurrencia de 22q11DS. Las células germinales masculinas continúan dividiéndose a lo largo de la vida, permitiendo la acumulación gradual de mutaciones, en contraste con las actividades de recombinación y reparación del ADN femenino, que están vinculadas a los oocitos detenidos [9]. En este escenario, se esperaría un aumento en el RR con la edad; sin embargo, esto no se observa, aunque este mecanismo no puede ser completamente excluido durante la gametogénesis.
No obstante, es más probable que tales alteraciones ocurran post-cigóticamente, durante el desarrollo embrionario o fetal, o incluso más tarde en la vida [9]. La NAHR somática es mediada por el intercambio intracromosómico durante la mitosis y puede ser responsable del mosaicismo somático [16].
¿Dónde ocurre el 22q11DS?
El síndrome se origina durante la gametogénesis o el desarrollo embrionario temprano. Como se mencionó, el principal mecanismo subyacente al 22q11DS de novo ocurre durante la meiosis, y por lo tanto durante la gametogénesis, ligeramente más frecuentemente en los oocitos. Aunque un solo evento meiótico no puede resultar en dos líneas celulares anormales, se ha propuesto un evento meiótico que causa duplicación seguido de una deleción mitótica en algunos casos [17]. Así, la presencia de una mutación de novo en mosaico implica un origen post-cigótico, que puede ocurrir durante el desarrollo embrionario o incluso postnatalmente. Estas mutaciones somáticas también se consideran de novo porque no son detectables en los padres de los individuos afectados [9] (Figura 3).
Tradicionalmente, el mosaicismo se ha considerado raro y se ha reportado principalmente en informes de casos o pequeñas series de casos. Sin embargo, un estudio reciente estimó una tasa de mosaicismo de deleción 22q11.2 en 28.2%. También observaron un bajo nivel (<35%) de líneas celulares eliminadas en más del 36% de los casos. Este hallazgo es importante para el asesoramiento genético, ya que uno de los padres puede tener un mosaicismo de bajo nivel en algunos o todos los tejidos [16].
Entonces, ¿cuáles son las consecuencias?
El 22q11DS puede afectar a todos los sistemas del cuerpo y puede ser potencialmente mortal en sus formas más severas. Se han reportado más de 180 características clínicas, asociadas con haploinsuficiencia de aproximadamente 90 genes identificados hasta la fecha. La cardiopatía congénita está presente en el 60–80% de los casos y varía en severidad, incluyendo una amplia gama de anomalías del arco aórtico y/o del tracto de salida cardíaco [1,18,19]. Esta también es la condición más común asociada con el paladar hendido, lo que puede llevar a insuficiencia velofaríngea y contribuir a problemas de habla y lenguaje. Las anomalías oftalmológicas también son comunes. El hipoparatiroidismo se diagnostica en el 40–75% de los casos y puede presentarse temprano en la vida como hipocalcemia que lleva a convulsiones. La inmunodeficiencia puede ocurrir en hasta el 80% de los casos, con una mayor susceptibilidad a trastornos autoinmunes [1]. Aunque la severidad de la inmunodeficiencia correlaciona con aplasia o hipoplasia tímica, el número de células T puede seguir siendo normal debido a tejido tímico ectópico dentro del mediastino [20].
La reducción del volumen cerebral (particularmente en los lóbulos temporales y el hipocampo) ocurre con la edad, y la reducción de la corteza prefrontal puede llevar a déficits en la memoria de trabajo. A menudo está presente la discapacidad intelectual, aunque más de la mitad de los individuos tienen inteligencia de límite a normal. Se estima que el 90–100% de los niños con síndrome de deleción 22q11.2 cumplen con los criterios para una discapacidad de aprendizaje. El fenotipo cognitivo también incluye un temperamento difícil y una predisposición a la ansiedad, la depresión y los déficits de atención. Hasta el 50% de los adolescentes reportan episodios psicóticos transitorios, mientras que aproximadamente el 33% de los adultos con VCFS son diagnosticados con esquizofrenia [1].
Las manifestaciones clínicas en la descendencia pueden resultar en un fenotipo cognitivo más severo. La razón de esta variabilidad en la expresividad clínica no se entiende bien.
Sin embargo, varios mecanismos biológicos parecen estar involucrados, incluyendo la sensibilidad a la dosis génica dentro de la región 22q11.2 y otros componentes hereditarios, como variantes en genes en el cromosoma 22q11.2 intacto y/o variantes modificadoras adicionales fuera de la región, que van desde genes que codifican proteínas hasta elementos reguladores [7]. Hasta la fecha, cinco genes dentro de la región 22q11.2 han sido principalmente implicados en el fenotipo neuropsiquiátrico: deshidrogenasa de prolina (PRODH), región crítica de DiGeorge 8 (DGCR8), catecol-O-metiltransferasa (COMT), T-box 1 (TBX1) y septina 5 (SEPT5). Los factores ambientales también pueden contribuir a estas manifestaciones [21].
Aparentemente, el nivel de mosaicismo no correlaciona directamente con el fenotipo. El mosaicismo de bajo grado (<35% de células con deleción) puede producir el fenotipo clásico, mientras que el mosaicismo de alto grado (>65%) puede resultar en presentaciones más leves. Esta diferencia puede deberse al mosaicismo específico de tejido [16].
En última instancia, el tamaño de la deleción y la ubicación genómica tienen un impacto directo en las manifestaciones clínicas. Los pacientes con deleciones anidadas proximales (que involucran LCR22A) comparten características fenotípicas mayores con aquellos que portan la deleción típica LCR22A–LCR22D. En contraste, las deleciones anidadas distales (LCR22B–LCR22D y LCR22C–LCR22D) están asociadas con fenotipos superpuestos pero menos penetrantes y son más frecuentemente heredadas [7].
Se han descrito deleciones atípicas, con puntos de ruptura ubicados entre las secuencias LCR22. La prevalencia de estas deleciones no estándar probablemente está subestimada porque son más difíciles de detectar con las pruebas genéticas actuales y están asociadas con fenotipos más leves [4].
¿Y entonces, qué podemos hacer al respecto?
La variabilidad de los síntomas en 22q11DS dificulta predecir la manifestación de la enfermedad [1]. Sin embargo, los problemas de salud asociados pueden impactar significativamente la vida diaria de la familia e influir en la toma de decisiones reproductivas. El diagnóstico genético preimplantacional y el diagnóstico prenatal pueden ofrecerse a parejas con mayor riesgo de recurrencia. Aunque hay una reticencia general a interrumpir embarazos afectados por 22q11DS, puede considerarse en regiones donde está permitido [2].
Las manifestaciones físicas a menudo son manejables. Las intervenciones quirúrgicas pueden corregir defectos cardíacos congénitos y paladar hendido, y en algunos casos, puede ser necesario el trasplante de timo debido a deficiencia de células T [1]. En cuanto a los resultados cognitivos, factores ambientales como la dinámica familiar, el estado socioeconómico y el estrés psicológico pueden influir en el perfil clínico, aunque se necesitan más estudios para aclarar estos efectos [21].
No sabemos hacia dónde nos llevará la ciencia, y muchas preguntas siguen sin respuesta: ¿Podemos prevenir las manifestaciones? ¿Cuál es el papel del mosaicismo somático? ¿Se encontrará una cura? ¿Podría la terapia génica dirigida a la causa subyacente proporcionar una cura? ¿O seremos capaces de modificar el curso de la enfermedad? ¿Podría eventualmente erradicarse esta condición?
La posibilidad de erradicar esta enfermedad es compleja porque la arquitectura genómica predispone a eventos recurrentes de 22q11DS. Sin embargo, aumentar nuestra comprensión de las funciones biológicas de los genes dentro de la región 22q11.2 y cómo los factores ambientales influyen en su expresión puede ayudar a aclarar la heterogeneidad de esta condición [21] y guiar futuras estrategias terapéuticas. Los avances continuos en el conocimiento, políticas de salud pública apropiadas y un diagnóstico preciso son esenciales para desentrañar la complejidad de esta y otras condiciones genómicas.
ABREVIACIONES
22q11DS: 22q11.2 deleción/Síndrome de DiGeorge; COMT: Catecol-O-metiltransferasa; DGCR8: Región Crítica de DiGeorge 8; LCRs: Repeticiones de baja copia; NAHR: Recombinación Homóloga No Alélica; PRODH: Prolina deshidrogenasa; RR: Riesgo de Recurrencia; SEPT5: Septina 5; TBX1: T-box 1.