
INTRODUCCIÓN
El mosaicismo diploide/triploide es una rara alteración genética caracterizada por la coexistencia de líneas celulares con diferentes números de cromosomas en el mismo individuo. Aunque la prevalencia exacta se desconoce debido a su rareza, comprender su impacto clínico y genético es esencial. Este trastorno presenta un amplio espectro de fenotipos clínicos, que van desde anomalías menores hasta condiciones severas que involucran crecimiento excesivo y asimetría corporal (Happle, 1987; Ledbetter et al., 1992).
La hemihipertrofia, una de las manifestaciones del mosaicismo diploide/triploide, es una condición en la que una parte del cuerpo es notablemente más grande que la otra, y está asociada con una variedad de síndromes genéticos (Hoyme et al., 1998). El diagnóstico temprano y el monitoreo son cruciales debido al mayor riesgo de ciertos tipos de cáncer en pacientes con hemihipertrofia (Clericuzio y Martin, 2009).
Los casos de mosaicismo diploide/triploide a menudo presentan desafíos diagnósticos, particularmente en el contexto prenatal. La presencia de una placenta molar, como se observó en este caso, puede ser un indicador temprano de esta condición (Vogel et al., 2010). La identificación precisa del mosaicismo es esencial para el manejo clínico y el asesoramiento genético, ya que tiene implicaciones significativas para el tratamiento y el pronóstico (Kalousek et al., 1993).
Este caso clínico destaca la importancia de la evaluación genética detallada en presencia de anomalías del desarrollo prenatal y postnatal. A través de un enfoque multidisciplinario que integra genética, ortopedia, radiología y pediatría, se puede lograr un diagnóstico preciso y un plan de manejo óptimo para estos pacientes complejos (Biesecker y Spinner, 2013).
METODOLOGIA
Los criterios de inclusión involucraron:
-Paciente con un fenotipo compatible con mosaicismo diploide/triploide.
-Evidencia de asimetría corporal y hemihipertrofia.
-Hallazgos prenatales sugestivos de anomalías cromosómicas.
-Confirmación de anomalías cromosómicas mediante pruebas genéticas.
Los criterios de exclusión involucraron:
-Casos con una etiología alternativa confirmada para la asimetría corporal.
-Pacientes con hallazgos clínicos no compatibles con mosaicismo diploide/triploide.
-Falta de consentimiento informado.
Descripción de las Pruebas Realizadas:
1. Secuenciación del Exoma Completo (WES):
o Fragmentación enzimática del ADN genómico.
o Enriquecimiento de la región objetivo usando sondas de captura.
o Secuenciación en una plataforma Illumina con >98% de cobertura del exoma humano.
o Análisis bioinformático para la identificación de variantes patogénicas.
2. Microarrays de SNP (Array-CGH):
o Uso del kit Infinium™ Global Diversity Array con Citogenética (Illumina).
o Evaluación de CNVs y aneuploidías en el genoma.
o Análisis usando el software NxClinical (BioDiscovery).
3. Análisis de Microsatélites (STR):
o Evaluación de 15 marcadores STR autosómicos y amelogenina.
o Uso del kit multiplex PowerPlex 16HS (Promega).
o Confirmación del origen parental del complemento cromosómico triploide.
4. Cariotipo en Sangre Periférica y Tejido Afectado:
o Cultivo de linfocitos para análisis citogenético.
o Evaluación de fibroblastos del tejido afectado para detectar mosaicismo.
o Confirmación de 46,XX en sangre periférica y 69,XXX en tejido afectado.
5. Pruebas de Imagen:
o Radiografía de la pierna derecha: Displasia ósea y crecimiento excesivo.
o Ultrasonido de tejidos blandos: Aumento del volumen de tejido en la extremidad inferior derecha.
o Doppler arterial y venoso: No hay anomalías vasculares.
o Ecocardiograma: Foramen oval permeable y derrame pericárdico mínimo.
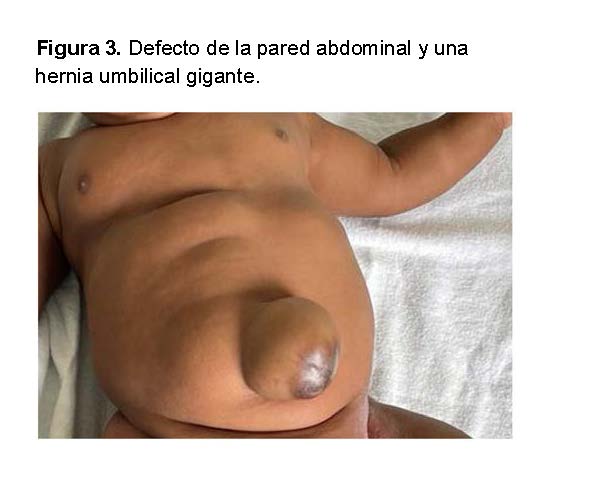
o Ultrasonido abdominal: Hernia umbilical gigante y lipohipertrofia.
Evaluaciones Clínicas Complementarias:
-Evaluación oftalmológica y auditiva: Hallazgos normales.
-Medición antropométrica y examen físico detallado.
Consideraciones Bioéticas: Declaramos haber obtenido el consentimiento informado del paciente para esta publicación, asegurando su comprensión de los objetivos, implicaciones y alcance de la divulgación de la información.
Esta publicación se adhiere a los principios éticos establecidos en la Declaración de Helsinki de la Asociación Médica Mundial, que rige la investigación que involucra a sujetos humanos, asegurando el respeto por la autonomía, confidencialidad y bienestar de los participantes.
Metodología de secuenciación
En la secuenciación del exoma completo, el ADN genómico fue fragmentado enzimáticamente y las regiones objetivo fueron enriquecidas usando sondas de captura de ADN. Estas regiones incluyen aproximadamente 41 Mb del exoma codificante humano (apuntando a >98% de las regiones codificantes de RefSeq del genoma humano construcción GRCh37/hg19), así como el genoma mitocondrial. La biblioteca generada fue secuenciada en una plataforma Illumina para lograr una profundidad de lectura de al menos 20x para >98% de las bases objetivo. Se aplicó un pipeline bioinformático interno, que incluye la alineación de las lecturas a la ensambladura del genoma GRCh37/hg19 y la Secuencia de Referencia de Cambridge revisada (rCRS) del ADN mitocondrial humano (NC_012920), llamada de variantes, anotación y filtrado exhaustivo de variantes. Se evaluaron todas las variantes con una frecuencia alélica menor (MAF) por debajo del 1% en la base de datos gnomAD y variantes causantes de enfermedades reportadas en HGMD®, ClinVar o CentoMD®. La investigación de variantes relevantes se centró en exones codificantes y ±10 nucleótidos intrónicos flanqueando genes con asociaciones claras gen-fenotipo (basado en información de OMIM®). Se consideraron todos los patrones de herencia potenciales. Además, se utilizó la historia familiar y la información clínica proporcionada para evaluar las variantes identificadas con respecto a su patogenicidad y causalidad de la enfermedad. Se establecieron criterios de calidad estrictos y procesos de validación para las variantes detectadas por NGS. Las variantes con baja calidad de secuenciación y/o cigocidad poco clara fueron confirmadas usando métodos ortogonales. En consecuencia, se garantiza una especificidad >99.9% para todas las variantes reportadas.
Para el análisis de arrays, se utilizó el kit Infinium™ Global Diversity Array con Citogenética (Illumina). Este kit contiene ~1.8 millones de marcadores SNP distribuidos a lo largo de todo el genoma, apuntando particularmente a más de 4800 genes asociados a enfermedades con 99.9% de cobertura exónica, permitiendo la detección de variaciones en el número de copias (CNVs). El ADN genómico fue amplificado, fragmentado e hibridado al array de acuerdo con las instrucciones del fabricante. Los resultados fueron analizados usando el software NxClinical (BioDiscovery). En el caso de los microsatélites, se analizaron 15 marcadores STR autosómicos y amelogenina usando el kit multiplex PowerPlex 16HS (Promega Corporation, Madison, USA).
RESULTADOS
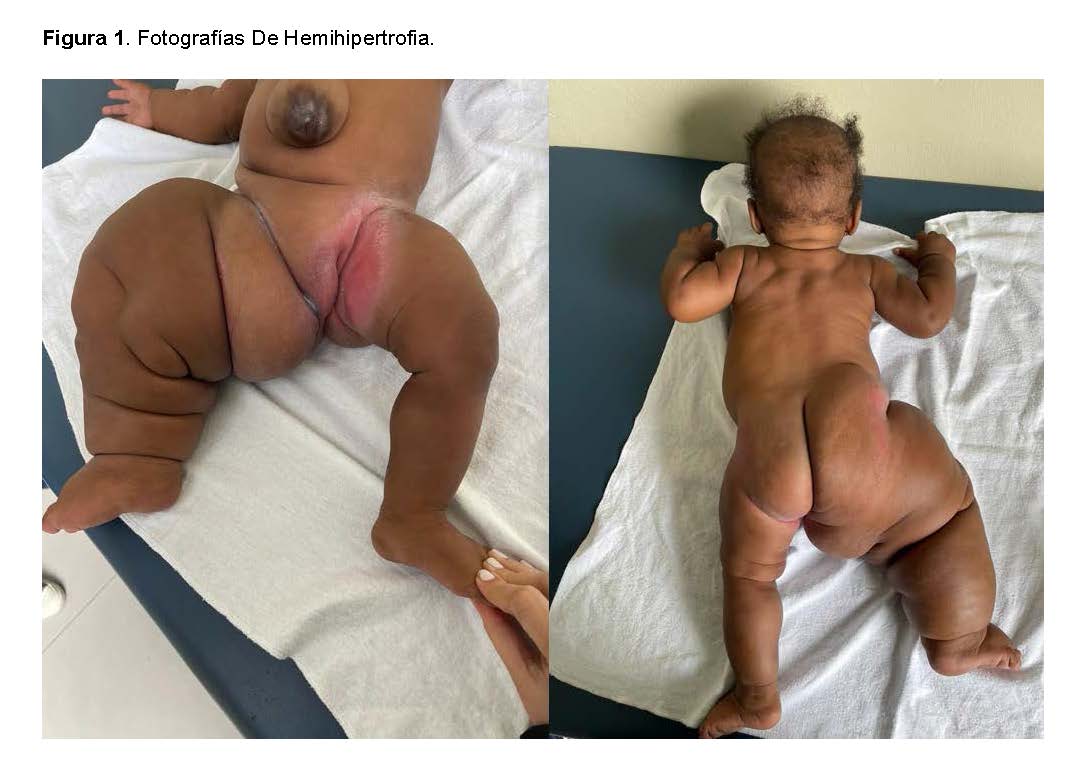
La paciente es una niña de un año, nacida de padres no consanguíneos: una madre de 20 años (G2P2C0A0) y un padre de 26 años. El seguimiento prenatal incluyó chequeos regulares, con la identificación a las 31 semanas de gestación de una anomalía placentaria caracterizada por molaridad o displasia mesenquimal, detectada mediante ecografía. Esta condición coexistía con un feto vivo que presentaba hipertrofia de la pierna derecha y displasia esquelética, así como polihidramnios leve. A las 36 semanas, se observó una discrepancia en el tamaño de la pierna fetal. La paciente nació a término mediante parto vaginal, pesando 6 libras, y fue dada de alta después de cinco días de observación sin incidentes.
La biopsia placentaria reveló una placenta monocoriónica monoamniótica madura con signos de anoxia focal, edema, hemorragia y corioamnionitis aguda focal. Además, se observaron cambios hidrópicos en las vellosidades, con formación de cisternas e hiperplasia trofoblástica leve, sugiriendo un componente molar parcial incompleto y la posibilidad de anomalías citogenéticas y displasia mesenquimal placentaria.
La paciente presentó retraso en el desarrollo motor y tiene un historial de alergia a la proteína de la leche de vaca. Ha sido hospitalizada dos veces debido a infecciones del tracto urinario. No hay antecedentes familiares reportados de relevancia médica.
Prueba Diagnóstica
Radiografía de la Pierna Derecha: Reveló acortamiento de la diáfisis y ensanchamiento de la epífisis, lo que sugiere displasia esquelética del miembro inferior derecho.
Ultrasonido de Tejidos Blandos: Detectó un aumento de tejido blando en el miembro inferior derecho.
Doppler Arterial y Venoso: No se evidenció patología vascular en el miembro.
Ecocardiograma: Identificó un foramen oval permeable y un derrame pericárdico mínimo.
Evaluaciones Oftalmológicas y Auditivas: Los resultados fueron normales.
Ecografía Abdominal: Reveló un defecto en la pared abdominal y una hernia umbilical gigante.
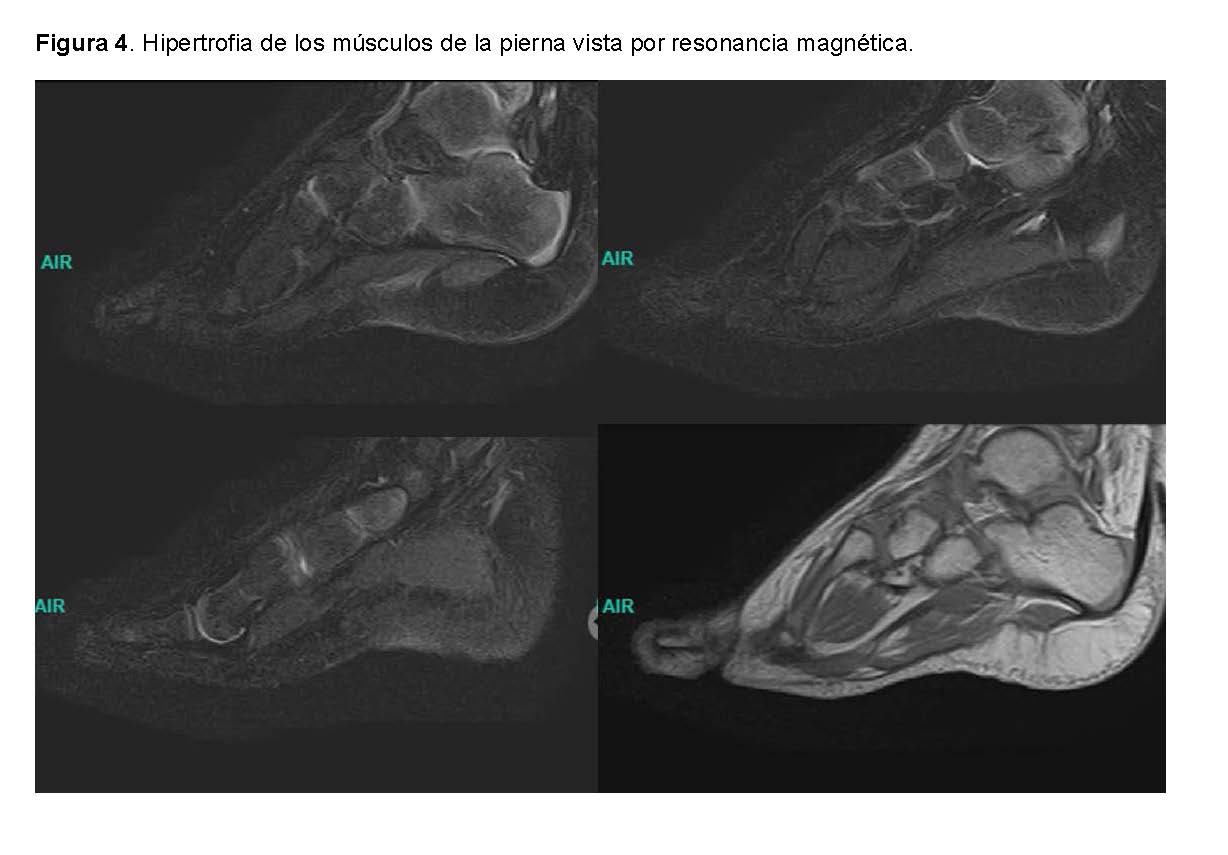
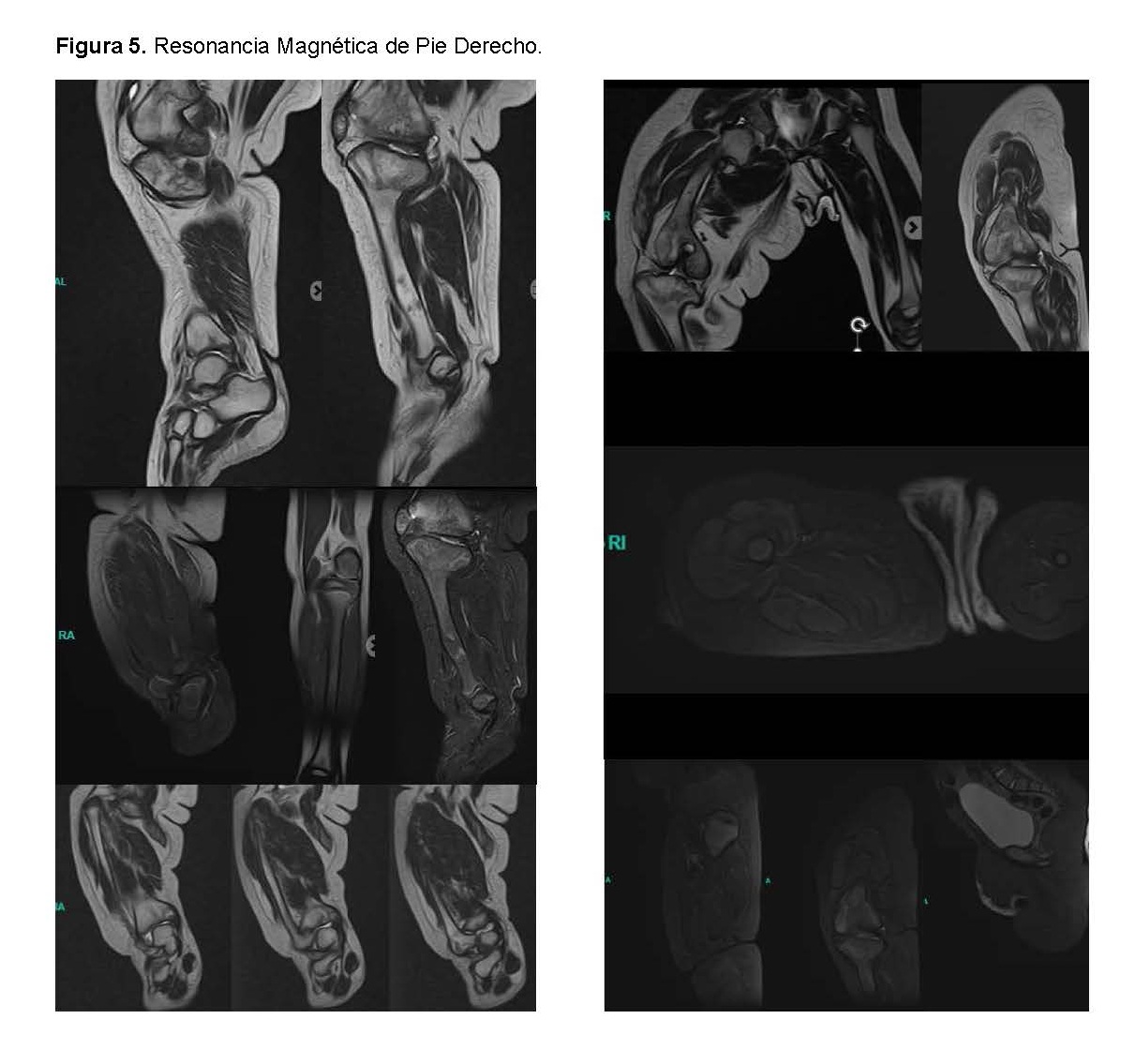
RM de la Pierna Derecha: mostró una Hiperplasia de la tibia con ausencia de la placa de crecimiento y distorsión morfológica con ensanchamiento del platillo tibial, una Hipertrofia del maléolo lateral con una apariencia anormal y una Hipertrofia de los músculos de la pierna excepto el gastrocnemio, que muestra atrofia y reemplazo graso.
RM del Muslo Derecho: Displasia de la cabeza femoral con hipoplasia de la diáfisis femoral. Probable insuficiencia acetabular basada en datos morfológicos. Fibrosis muscular con reemplazo graso de las fibras musculares del cuádriceps. Hipertrofia muscular generalizada.
Resonancia Magnética del Pie Derecho: La resonancia magnética mostró una divergencia calcáneo-astragalina y acortamiento del tendón de Aquiles.
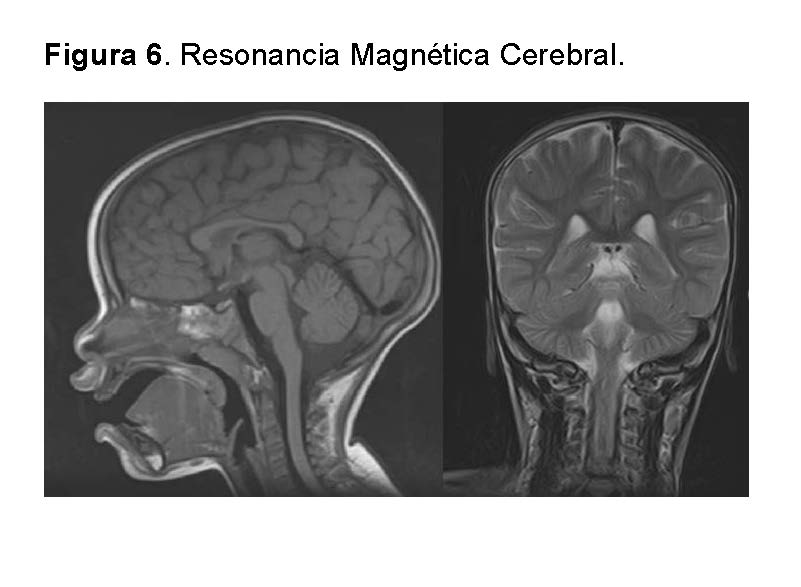
Resonancia Magnética Cerebral: La resonancia magnética cerebral no mostró evidencia de anomalías estructurales, lesiones, masas, signos de infección o inflamación, ni anomalías vasculares. La morfología y la señal de las estructuras intracraneales eran normales, incluyendo la sustancia blanca y gris, los ventrículos y los espacios subaracnoideos. No se observaron signos de edema cerebral ni desplazamiento de la línea media. El estudio es consistente con un examen neurológico sin hallazgos patológicos.
Las medidas antropométricas se describen a continuación:
-Peso: 9 kg (percentil 50)
-Altura: 85 cm (>percentil 97)
-Circunferencia de la cabeza: 44.8 cm (percentil 25)
Durante el examen físico, se observaron asimetría craneal, línea de cabello alta, prominencia frontal, epicanto, telecanto, puente nasal aplanado y otras características dismórficas. La pierna derecha está significativamente agrandada con pliegues cutáneos alterados, sugiriendo un síndrome de sobrecrecimiento con asimetría corporal.
Durante la evaluación genética del tejido afectado, el análisis de ADN (pierna derecha con hemihipertrofia) mostró un complemento cromosómico triploide: arr(X,1-22)x3. El análisis de fragmentos de marcadores microsatélites reveló un patrón trisómico dialélico (2:1) para 11 STRs informativos. Los datos de NGS y el perfil de SNP del análisis de microarray (utilizado como control interno) fueron consistentes con un genoma triploide (69,XXX). La anomalía cromosómica se clasifica como patogénica según las directrices de ACMG.
En el análisis de ADN de sangre periférica, no se detectaron variantes de secuencia de ADN clínicamente relevantes, variaciones en el número de copias u otros hallazgos asociados con el fenotipo reportado en la muestra de ADN de sangre periférica. La secuenciación del exoma mostró un complemento cromosómico diploide femenino normal, confirmado por microarray cromosómico y consistente con el cariotipo previo del paciente: arr(X,1-22)x2 htz.
Resultados del cariotipo: La prueba de sangre periférica mostró un cariotipo femenino normal de 46XX. Sin embargo, la pierna derecha tiene un cariotipo anormal de una triploidía de 69 XXY.


DISCUSION
Este caso de mosaicismo diploide/triploide representa el primer informe documentado en la República Dominicana, destacando la importancia de su descripción detallada y análisis. El paciente presenta un fenotipo caracterizado por hemihipertrofia, sobrecrecimiento y anomalías esqueléticas, hallazgos que se alinean con los reportados en estudios previos sobre esta condición (Happle, 1987; Ledbetter et al., 1992; Hoyme et al., 1998). Sin embargo, la combinación de estas características con antecedentes prenatales de displasia placentaria y anomalías vasculares refuerza la heterogeneidad fenotípica del mosaicismo diploide/triploide.
Los hallazgos genéticos en este paciente enfatizan la importancia de analizar muestras del tejido afectado, ya que el cariotipo de sangre periférica era normal (46,XX), mientras que la triploidía (69,XXX) fue confirmada en el tejido de la pierna hemihipertrofiada. Este hallazgo es consistente con informes previos donde la sangre periférica no siempre refleja la presencia de mosaicismo (Van De Laar et al., 2002; Boonen et al., 2011). Además, el estudio de microsatélites confirmó un patrón trisómico dialélico (2:1), indicando que el conjunto extra de cromosomas es de origen materno, en línea con la hipótesis de inclusión del segundo cuerpo polar durante la embriogénesis (Brems et al., 2003).
Un análisis comparativo con la literatura médica revela varias similitudes y diferencias fenotípicas. En estudios previos, el mosaicismo diploide/triploide se ha asociado con retraso en el crecimiento, hipotonía, anomalías faciales y displasias esqueléticas (Graham et al., 1981; Rittinger et al., 2008). En nuestro caso, se identificaron anomalías similares, aunque el paciente presenta un fenotipo de sobrecrecimiento más pronunciado, lo que podría estar relacionado con la distribución específica de tejido de las células triploides. Esto se alinea con estudios que sugieren que la expresión fenotípica depende del porcentaje de células triploides en tejidos específicos (Oktem et al., 2007).
Desde un punto de vista clínico, el seguimiento a largo plazo es esencial. Se ha descrito un aumento del riesgo de neoplasia en pacientes con hemihipertrofia, particularmente en aquellos con alteraciones genéticas subyacentes (Clericuzio & Martin, 2009). En este contexto, el paciente requiere un monitoreo periódico mediante ecografía abdominal y pruebas adicionales para la detección temprana de tumores asociados. La literatura respalda este enfoque, enfatizando la necesidad de evaluaciones regulares y seguimiento multidisciplinario (Biesecker & Spinner, 2013).
Desde una perspectiva bioética, el manejo de pacientes con mosaicismo diploide/triploide presenta desafíos en el asesoramiento genético, ya que su expresión clínica puede ser muy variable. En nuestro caso, se proporcionó información detallada a la familia sobre el pronóstico incierto y la necesidad de estudios genéticos en muestras de tejido afectado para una evaluación más precisa. Esto es consistente con informes que destacan la importancia del asesoramiento genético en condiciones raras y la necesidad de un manejo individualizado (Kalousek et al., 1993; Vogel et al., 2010).
Este caso contribuye significativamente a la comprensión del mosaicismo diploide/triploide, enfatizando la importancia del análisis genético en el tejido afectado para una caracterización precisa. El fenotipo observado confirma la variabilidad clínica de la condición y refuerza la necesidad de un enfoque multidisciplinario en el manejo de estos pacientes. La vigilancia periódica y el seguimiento genético siguen siendo aspectos esenciales para asegurar una atención óptima y personalizada.
Agradecimientos
A Jessie PEPÉN BODRÉ.