INTRODUCTION
Craniofacial anomalies are a group of rare congenital malformations, with prevalence of 88.4 per 100,000 births and various etiologies. A commonly diagnosed syndrome with craniofacial features is craniofacial microsomia [1].
Craniofacial microsomia (CFM; OMIM#164210) is the current form of designation for a condition that received different names and characterizations over the years. Whether called by CFM, Goldenhar syndrome [2], Oculo-auriculo-vertebral spectrum [3] or Hemifacial Microsomia [4], it is known that this condition consists of an asymmetric congenital malformation of the first and second branchial arches, possibly due to vascular injury or altered migration of neural crest cells [5]. Thus, all structures derived from these arches may be affected resulting in a large spectrum of clinical features, encompassing ears, eyes, muscles, facial bones, and cervical vertebrae [6].
The broad phenotype spectrum urges physicians to consider other syndromes with craniofacial abnormalities such as Treacher Collins syndrome, branchio-oto-renal syndrome, maxillofacial dysostosis and Cat-Eye Syndrome [7].
The Cat-Eye Syndrome (CES; OMIM#115470), occurs due to a partial tetrasomy of chromosome 22, resulting from a dicentric supernumerary marker with satellites at both its ends, inv dup(22) (pter->q11.2::q11.2->pter) [8,9]. This chromosome arm has 8 regions of low copy repeats (LCR) called LCR22A to LCR22H, and its homology predisposes to the formation of rearrangements [10]. CES is clinically characterized by a very variable phenotype with the presence of multiple malformations, mainly affecting the eyes, ears, anorectal and urogenital regions [8]. The genomic region related to this comorbidity is mapped between the centromere and the LCR22A and named Cat Eye Syndrome Critical Region (CESCR) [11]. There are two types of CES: type I is characterized by a breakpoint on or before LCR22A, and type II includes the DiGeorge syndrome (DGS) critical region [12].
Here we report a patient clinically misdiagnosed with CFM, following the CARE checklist [13], who has laboratory tests compatible with type II CES. In order to discuss our findings, we performed a literature review. As hers maker's chromosomes diverged slightly with the ones found recently published, we expanded it to 20 years of case and series reports of patients diagnosed with CES, which highlight the rarity of our case. We also discussed and proposed a mechanism for this complex chromosome alteration formation.
Lastly, we briefly reported the phenotype of previously unpublished cases in Brazil and examined the epidemiological and clinical concerns.
CASE REPORT
The patient is a female, the first child of a young, healthy, non-consanguineous couple from the south region of Brazil. The pregnancy was uneventful, and there were no cases of facial dysmorphisms in the family history (three generation pedigree). She was delivered at 35 weeks and 1 day, with a low birth weight (1920 grams; below the 10th percentile). In the delivery room, the pediatrician reported ear abnormalities. No abdominal abnormalities were reported at physical examination and ultrasound.
A CT scan at 1 month revealed right mandibular hypoplasia and ear canal abnormalities, including possible malleus-incus fusion, and dislocation of these bones toward the upper lateral tympanic membrane. Brainstem auditory evoked response testing also showed dysfunction in the right ear.
At 2 years old, the patient developed recurrent urinary tract infections and had febrile seizures on three occasions. The Tc-99m dimercaptosuccinic acid renal scan was normal, and no anatomical anomalies were observed on ultrasound; antibiotic prophylaxis was prescribed. An EKG showed a shortened PR interval (0,09s; HP: 0005165), but without clinical repercussions or echography abnormalities.
At 3 years old, the patient was referred to the medical genetics outpatient service and evaluated by an experienced dysmorphologist. Motor neurodevelopmental delay was detected. She had a low weight (below the 5th percentile; HP: 0004325), curly hair (HP: 0002212), right microtia (HP: 0008551), right microphthalmia (HP: 0000568), right epicanthic fold (HP: 0000286), wide nasal bridge (HP: 0000431), high palate (HP: 0000218), right preauricular skin tag (HP: 0000384) and pit (HP: 0100277), and sacral dimple (HP: 0000960). Epibulbar dermoids, oral cleft or any other physical abnormalities weren’t noted, and ophthalmological evaluation was uneventful. As the patients had asymmetrical facial dysmorphisms, the hypothesis of CFM was proposed.
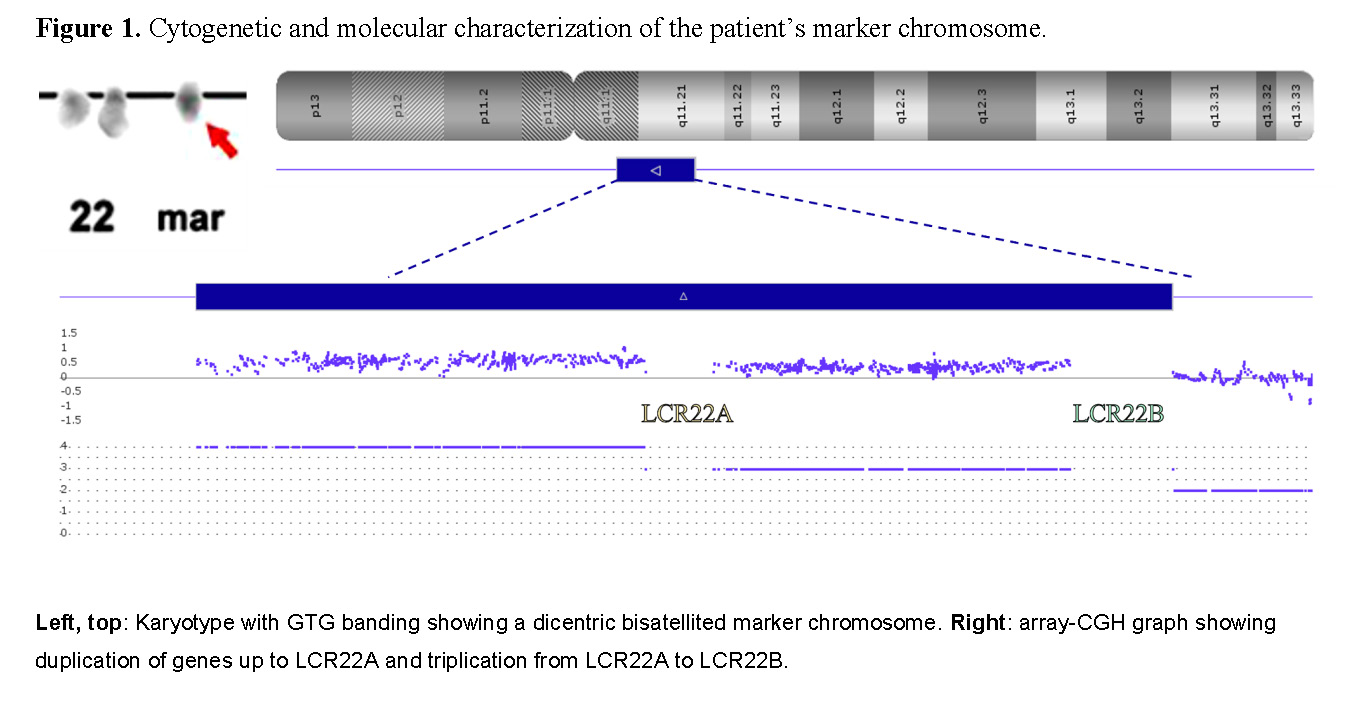
A G-banding karyotype was performed, identifying 47,XX,+mar[25]. The array-based comparative genomic hybridization (array-CGH - CytoScan™ 750K Suite - Thermo Fisher Scientific, Catalog #901859) confirmed that the extranumerary marker derived from a chromosome 22 and delimited a LCR22B breakpoint (Figure 1). Thus, the asymmetrical type II CES was confirmed, and the complete cytogenetic result was defined as 47,XX,+mar[25]. arr[GRCh37] 22q11.1q11.21(16888899_18644240x4,18649189_20716903x3,20723685x2).
At 10 years old, the patient was also diagnosed with Attention-deficit/hyperactivity disorder.
DISCUSSION
Our original clinical hypothesis was CFM, but the cytogenetic and molecular findings established the diagnosis of CES. In an attempt to determine the patient’s diagnosis, we come across the similarity of clinical manifestation of both syndromes – mainly involving preauricular pits, microtia, and asymmetrical facial dysmorphisms.
The classical triad of CES is preauricular anomalies, anal malformation and iris coloboma. So, our patient has just one classic feature (pre auricular abnormality). In a cohort with patients with gain in the region LCR22A most individuals have only one or two features described on the classic triad (75%) [12]. Therefore, it may not be useful to identify most cases of CES.
As well, considering the CFM diagnostic criteria (FACIAL and ICHOM criteria described by Renkema et al.) [14], our patient could be wrongly diagnosed with CFM if not identified the cytogenetic gain/identification of other syndromes, established as exclusion criteria.
In consequence of this clinical overlap, it is not unusual to find individuals with CES and clinical diagnosis of CFM. In literature, we can find some reports, including a recent study conducted by Spineli-Silva et al. [6], which explored the relationship between the two phenotypes based on case reports of three patients. So, even if CFM is considered by some authors [14], CES must be considered as a differential diagnosis.
This phenotypic overlap and the need for cytogenetic analysis may be a significant diagnostic barrier in an underdeveloped and emerging country like Brazil. According to the Brazilian Rare Diseases Network (BRDN; a population database of rare diseases), 19 patients with possible CFM are assisted by clinical geneticists, and 20 patients have been confirmed with this condition. None of them reported cytogenetic and/or molecular tests, even though the FACIAL and ICHOM criteria suggest that other syndromes must be ruled out. In addition, only five other patients are reported to have a confirmed CES diagnosis.
It must be highlighted that, although the institutions that compose the BRDN provide care for the majority of the Brazilian population, they provide, on average, 84 microarray and 47 fluorescence in situ hybridization (FISH) tests per month [15]. Thus, the low capacity may explain the incomplete investigation of most cases.
Among the patients diagnosed with CES, only one had the classic triad, one had two of the signs (anal anomaly plus coloboma), and the remaining three had none. The clinical manifestations of these patients can be seen in Table 1; all cases were de novo (non-familial recurrence in a three-generation pedigree).
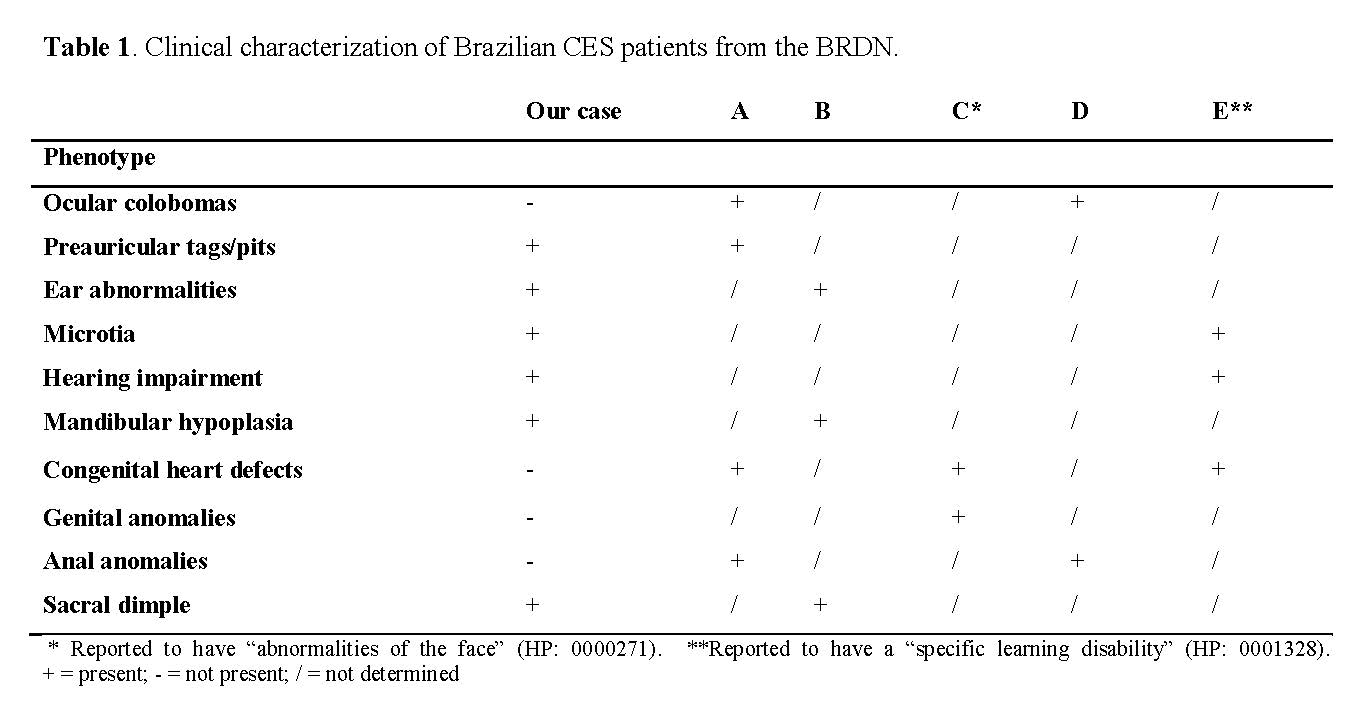
Although we do not have enough information to determine the type of CES with which these patients had been diagnosed, we can infer from the broad phenotype spectrum that clinical evaluation is insufficient to establish the diagnosis, as previously reported in the literature [12]. Therefore, laboratory findings are essential to confirm the diagnosis.
Since facial dysmorphisms can be observed from birth and few cases are identified prenatally (0.6% [1]), we believe pediatricians and neonatologists should be able to identify possible cases of CFM, CES, and other syndromes with craniofacial abnormalities, and properly refer these patients to clinical geneticists, who will be able to provide genetic counseling.
This challenge relates to the Brazilian National Policy for Comprehensive Care in Clinical Genetics. Even though it has been established for more than a decade, the public health system still lacks clinical pathways to investigate and manage rare diseases. Additionally, this legislative ordinance was not followed by an executive one; therefore, there is no national, consolidated, long-term financial plan [16].
These political and administrative issues, in addition to the variable phenotype previously discussed, may explain the long patient odyssey of 16 years.
Literature review
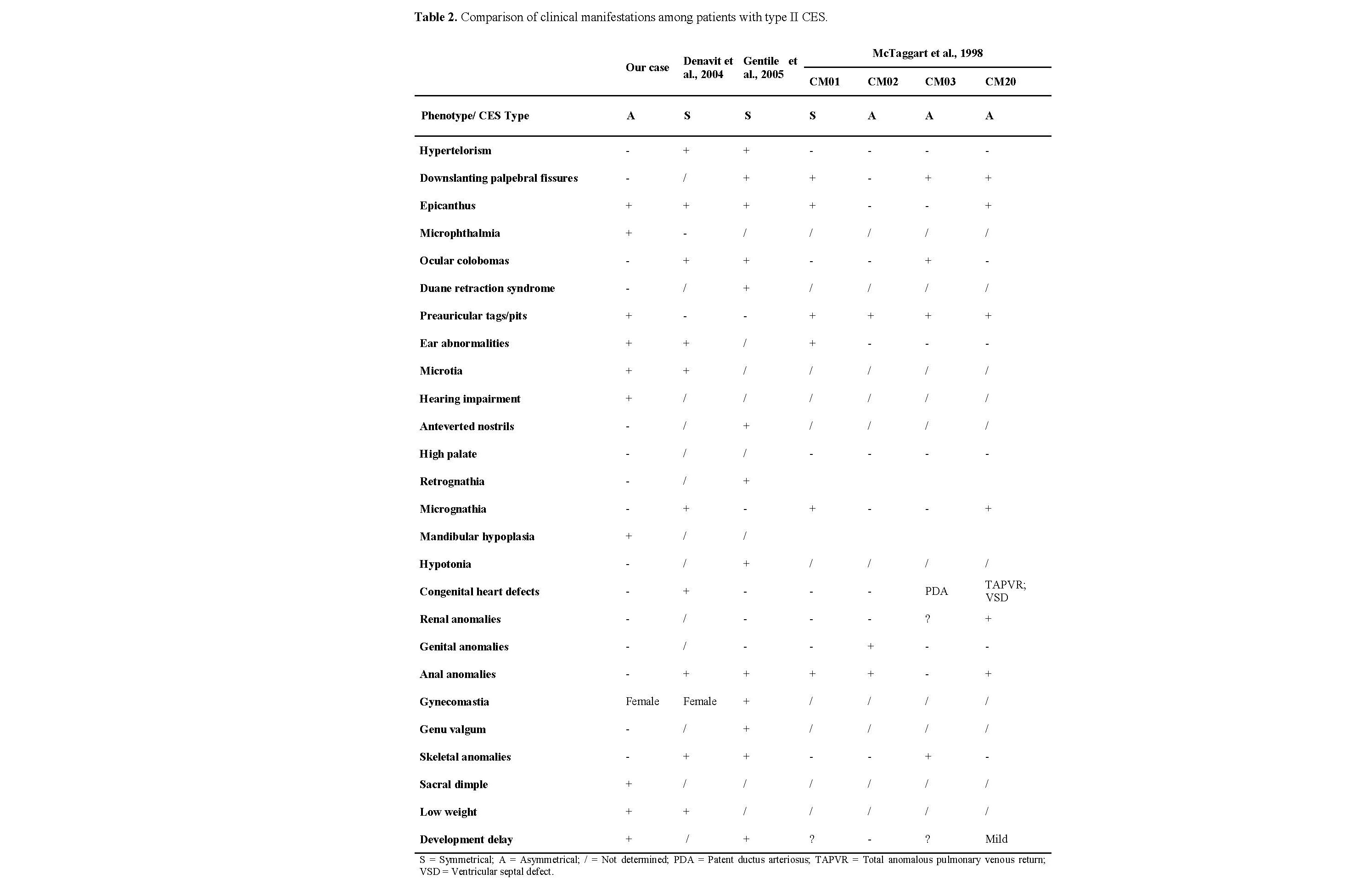
Our case has a duplication ranging from pter to LCR22B. When compared with recent research on CES, all reported cases [6] have the duplicated portion ranging from pter to LCR22A (which includes the genes up to USP18). Despite this difference in loci (which involves 30 genes), no significant phenotype variation was observed.
Thus, we decided to expand our literature search, seeking CES patients with molecular characterization. In the last 20 years, a total of 53 cases [17–47] have been reported, and only two were found to carry a larger marker chromosome; however, unlike our report, both were symmetrical.
The first one shows severe craniofacial alterations, including absence of both ears [47]. The second [45] manifests Duane retraction syndrome, a finding reported in six other cases of CES, but in type I [43,44,32,29,31].
The clinical manifestations of these patients are compared with our patient in Table 2. We also compared the morphological findings with those in the first case series [48], adding four more cases of type II CES to our set.
Critical Cat Eye Syndrome Chromosome Region
The Cat Eye Syndrome Chromosome Region (CESCR) is mapped between the centromere of chromosome 22 and LCR22A [genomic coordinates (GRCh38/hg38): 22:17,359,949–17,558,151]. This explains the clinical manifestations of those patients reported with the breakpoint on or before this region (Supplemental Table 1). To date, 14 genes have been identified in the CESCR [23], and individuals with intrachromosomal duplications or triplications of this region have shown phenotypes like CES, as well as ring chromosomes [49–53,39], indicating that partial trisomy is sufficient to cause the major and minor features of CES, independently of the chromosomal extranumerary marker structure [54]. Even then, there are still some clinically defined or convincing CES cases whose genetic etiology remains to be elucidated [12].
Unlike trisomy, haploinsufficiency of these regions appears to be harmless [55], as trisomy of the genes after LCR22A does not appear to produce a phenotype more severe than that associated with type I CES. Therefore, determination of the type of CES breakpoint would not have prognostic value [54]. But is that really the case?
Although it has been suggested that the inclusion of genes after LCR22A does not contribute to the phenotype of CES, duplications within this region were described in three patients with a similar clinical outcome: bladder exstrophy. The first case, a non-syndromic child with classic bladder exstrophy, had a duplication ranging from CLTCL1 to IZTR1 genes, indicating an LCR22A–LCR22D rearrangement [56]. The other two cases were unrelated patients with the same outcome plus hearing impairment, with duplicated regions described as arr.[NCBI36/hg18]17313883_19854524 in case 1 and arr.[NCBI36/hg18]17313883_20006849 in case 2, both also rearrangements involving LCR22A–LCR22D [57]. The proposed mechanism for bladder exstrophy involves DGCR8 overexpression interfering with p63 regulation through microRNA processing [56]. However, our patient has DGCR8 duplicated and does not show any sign of bladder exstrophy, indicating that the genomic contribution to this outcome likely lies beyond LCR22B. Considering other urinary anomalies, about 8% of patients with CES may have vesicoureteral reflux [12], but there is still no satisfactory explanation for this association.
Intriguingly, copy-number variation (CNV) gain of the DiGeorge region may have some clinical impact after all. This condition is known as 22q11.2 duplication syndrome (OMIM #608363), and its phenotypic features overlap with those of DiGeorge syndrome (22q11.2 deletion). The main gene related to this condition is TBX1, a member of the T-box gene family, whose over- or underexpression has been implicated in the full spectrum of DiGeorge syndrome malformations [56]. Still, TBX1, together with CLTCL1, GSC2, HIRA, and MAPK1, forms a group of genes suggested to be candidates for involvement in the asymmetric nature of craniofacial microsomia etiology [58]. All these genes are located between LCR22A and LCR22B. Consequently, they are duplicated in our patient but not duplicated in the most common cases of CES, namely type I CES.
Another study investigating microduplications on chromosome 22q11.2 analyzed 31 cases, including three with duplications from LCR22A to LCR22B, four from LCR22B to LCR22D, five from LCR22C to LCR22D, sixteen from LCR22A to LCR22D, and three in distal LCR regions (one LCR22E–LCR22F and two LCR22E–LCR22H). Interestingly, individuals with duplications involving LCR22A–B and LCR22B–D showed normal development on physical examination; however, renal abnormalities were documented in three fetuses, all involving LCR22D, suggesting that duplications in this region could play a pivotal role in kidney phenotypes. Additionally, rare phenotypes such as skeletal abnormalities, facial anomalies, and thymic hypoplasia were observed, indicating that gene dosage alterations in the 22q11.2 region are critical determinants of clinical manifestations [59]. These variations in clinical outcomes may reflect the influence of genomic configuration or the positional effects of genes on phenotype.
Duplication mechanisms
The most accepted mechanisms implicated in CES rearrangements rely on homologous recombination (HR) and non-allelic homologous recombination (NAHR) events between the LCR22s during meiosis, as well as duplications and deletions within the 22q11.2 region [60,61,20]. The reason for such a multitude of rearrangements lies in the complicated inverted and direct orientation of sequences in the LCR22s [60]. However, the formation of a bisatellited marker and interchromosomal duplication structures is slightly different. CES type I and type II symmetrical markers result from a correct pairing of homologous chromosome or sister chromatid regions followed by an erroneous resolution, while CES type II asymmetric and interchromosomal duplications result from NAHR, which means that the pairing occurs between different LCRs (Figure 2A). One way to see this is that the interchromosomal duplications are type II asymmetric recombination followed by a correct resolution. In those cases, usually only one of the two centromeres is functional and is randomly pulled by the spindle fiber [20].
Theoretically, the rearrangement could occur between any LCR22, but in our review, we have not come across any case beyond LCR22B in a bisatellited chromosome marker. The interchromosomal duplication, on the other hand, has been described with duplicated genes, either in direct or inverted orientation, throughout the long arm of chromosome 22.
One particular DNA structural element, a palindromic AT-rich repeat (PATRR), manifests size polymorphisms and is present within LCR22A, LCR22B, and LCR22D, surrounded by a subtype of human satellite I (HSAT I) and an AluSc element, forming a 2.4-kb tripartite structure [62]. The symmetry and length of the PATRRs greatly influence the frequency of de novo translocations, due to their propensity to form a cruciform DNA conformation in vivo, which may induce breakage and consequently illegitimate joining [63]. These PATRRs have been suggested to be involved in reciprocal translocations including the recurrent rearrangements t(11;22)(q23;q11.2), t(17;22)(q11.2;q11.2), and t(8;22)(24.1;q11.2), as well as non-recurrent events such as t(4;22)(q35.1;q11.2) and t(1;22)(p21.1;q11.2) [64]. LCR22A and LCR22D are the largest LCR regions, which may explain their higher rearrangement frequency. As can be observed, type I CES is by far the most common. However, most of the aforementioned translocations involve LCR22B, which makes this hypothesis less probable.
An intriguing fact is that HR or NAHR between homologous chromosomes would not result in a normal chromosome 22 able to segregate. Thus, in this scenario, it would not be possible to obtain a karyotype with an extranumerary bisatellited marker. Rearrangements between sister chromatids, on the other hand, would allow it. But is there another option?
Hairpins formed on single-stranded DNA ends exposed by resection can inhibit efficient DNA repair [65]. Another explanation that comes to mind involves not a break but a collapse of the replication fork. In this scenario, a regressed fork promotes an extra copy and forms a Holliday junction, in which the repair process generates not only the bisatellited marker but also a normal chromosome 22 (Figure 2B–C). According to replication-based mechanisms, this juxtaposition of non-contiguous DNA sequences can range from a few hundred base pairs to megabases and may be driven by as little as two to six nucleotides of homology [66].
This mechanism could be a plausible explanation for those cases of CES in which the breakpoint occurs before LCR22A, as well as symmetric type I and II CES, since the collapse may be caused by a non-B DNA configuration of PATRR (Figure 2B). Asymmetric CES could also arise, but with an additional step of template exchange to a proximal replication fork, which in this case would be located near LCR22B (Figure 2C).
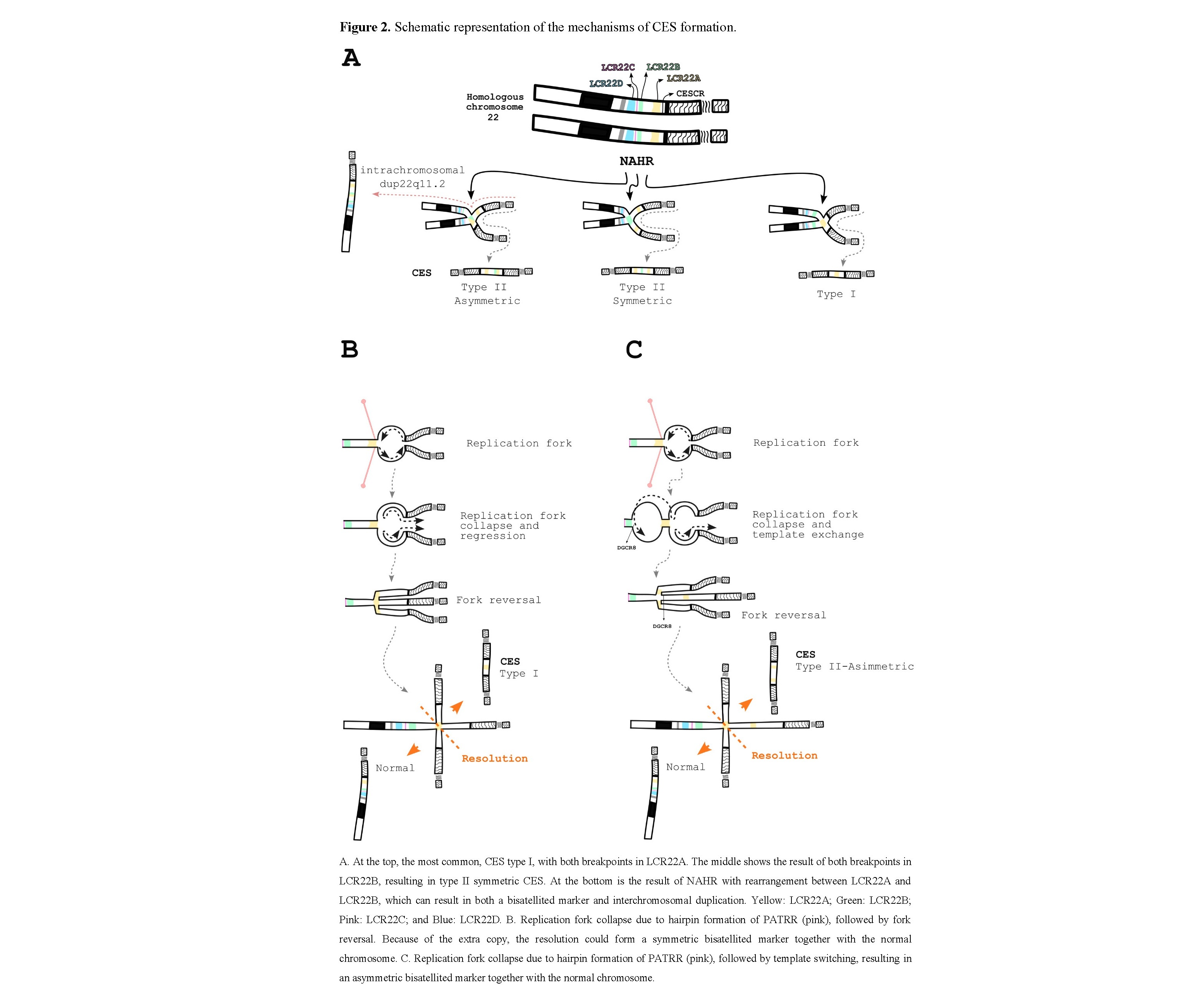
Regardless, differences in chromosome pairing can lead to differences in genetic content. In cases of symmetric CES, all genes on the marker chromosome will be quadruplicated, with one set in an inverted configuration. Meanwhile, asymmetric type II CES will have only the genes up to LCR22A quadruplicated and those from LCR22A to LCR22B triplicated, also with one set in an inverted configuration. These variations in genetic content and gene orientation may lead to different combinations of gene expression that could influence clinical manifestations.
CONCLUSION
We reported a case of CES and reflected on the clinical, epidemiological, and molecular aspects of this disease. As only a minority of patients present the classic triad, CES must be considered a differential diagnosis of CFM. In addition, this clinical overlap may impair diagnosis in underdeveloped countries, where laboratory investigations are essential to confirm CES.
Answering our main question, it appears that, at least in this case, size really does not matter. This suggests that an increase in copy number variation (CNV) encompassing the whole set of genes between LCR22A and LCR22B, as suggested by other reports, does not play a major role in the clinical manifestations of CES. However, if these genes are supposedly implicated in CFM and other phenotypic manifestations, why does their overexpression not translate into clinical findings? One possible explanation could lie in the genomic configuration or the positional context of the genes within the chromosome.
The classical definition of CES infers the presence of a chromosomal marker structure, but more recent findings regarding CNVs within the CESCR [49–53,39] suggest that the structure itself may not be the determining factor. Despite this, an important question remains: what path should be taken to better understand the disease etiology?
Contrary to what has been previously proposed, our findings suggest that the most likely mechanism may be replication-based, since such a mechanism could generate both chromosomes simultaneously: a normal chromosome 22 and a bisatellited marker chromosome. It would therefore be highly informative to investigate the breakpoints and genetic content of bisatellited markers in detail, as demonstrated in our case using a TXNRD2 probe. By doing so, it may be possible to extrapolate these findings to other disorders caused by similar chromosomal structures, such as inv dup(15) syndrome, ultimately improving patient care through more accurate genotype–phenotype correlations and better genetic counseling.
Acknowledgements
We would like to thank the patient and her family. This study was supported by InRaras - National Institute of Rare Diseases.
Author contribution statement
RM: Conceptualization, Data curation, Writing – original draft; JPPT: Conceptualization, Data curation, Writing – original draft; MM: Data curation, Writing – original draft; LDW: Data curation, Writing – original draft; GA: Formal analysis, Methodology, Writing – review & editing; PT: Validation, Writing – review & editing; BMO: Supervision, Writing – review & editing; TMF: Supervision, Writing – review & editing; PRGZ: Supervision, Project administration, Writing – review & editing.