
INTRODUCTION
The 22q11.2 deletion, also known as DiGeorge syndrome (22q11DS, OMIM #192430, #188400), is a multisystem disorder that can lead to a broad range of clinical manifestations associated with congenital heart disease, cleft palate, endocrinopathies, seizures, immunodeficiencies, intellectual disability, and neuropsychiatric features [1,2].
This deletion affects approximately 1 in 2,148 live births, is estimated in 1 in 992 unselected pregnancies, and 1 in 1,497 miscarriages, making it one of the most common genetic disorders [3]. The origin of this disease is very complex, and to better understand it, we address here some crucial questions.
Why does 22q11DS happen?
There are eight low copy repeat (LCR) clusters spread along the long arm of chromosome 22. They are denoted as LCR22A to LCR22H [4] and originated at least 40 million years ago [5] through genome shuffling via Alu-mediated rearrangements [6]. The presence of these LCRs makes this region highly unstable, which is consistent with the high incidence of this condition.
The typical deletion involves LCR22A and LCR22D (3 Mb) and occurs in 85% of individuals. When LCR22F–LCR22B and/or LCR22C are involved, the deletions are defined as “nested.” Around 5% of affected individuals carry the LCR22A–LCR22B (1.5 Mb) deletion, 2% LCR22A–LCR22C (2 Mb), 4% LCR22B–LCR22D (1.5 Mb), and 1% LCR22C–LCR22D (1 Mb) (Figure 1). Deletions involving LCR22E to LCR22H are less frequent, with phenotypes differing from 22q11.2DS [4,7]; therefore, we will focus only on LCR22A to LCR22D deletions.
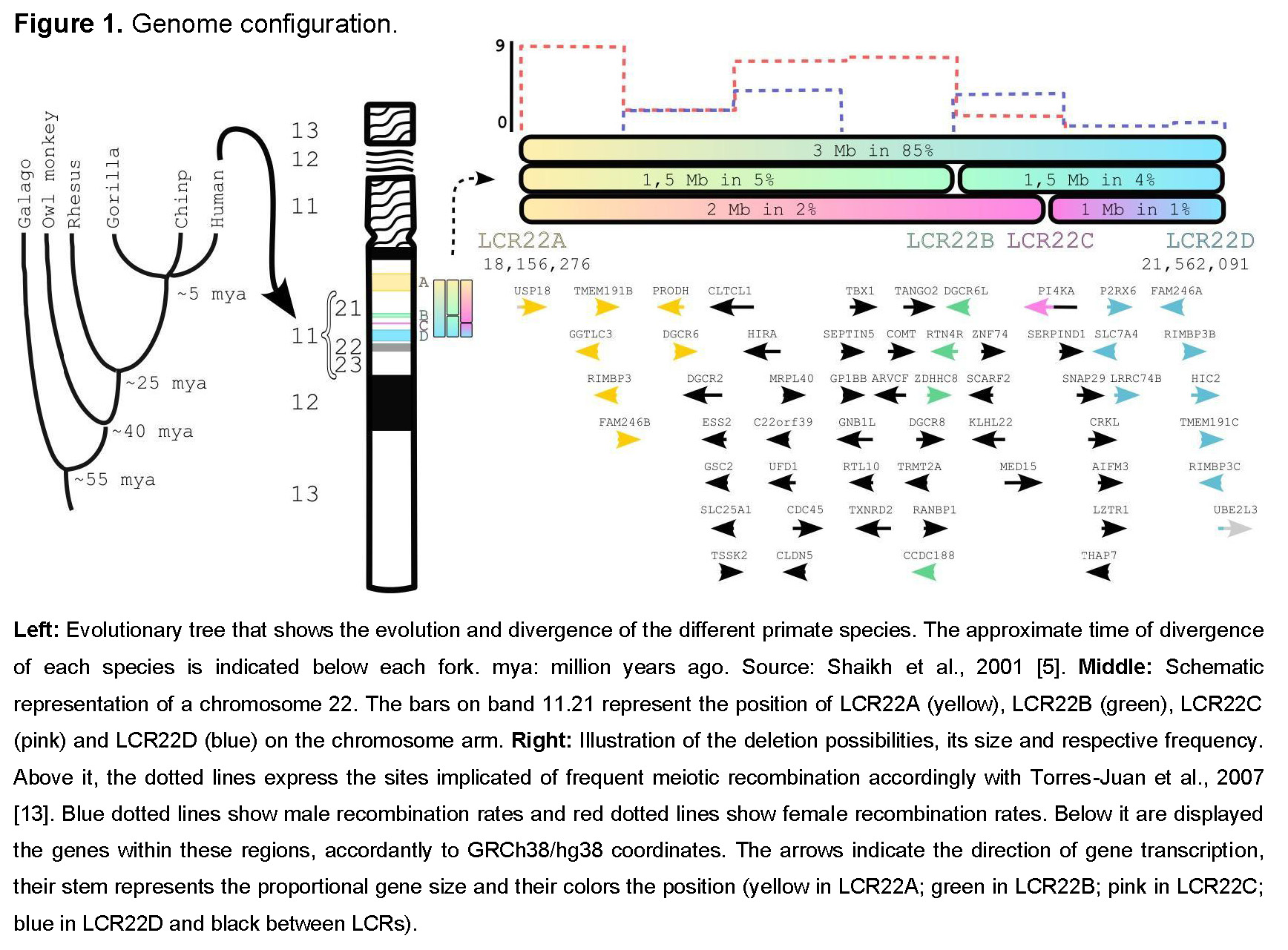
How is it generated?
It is generated through interchromosomal or intrachromosomal nonallelic homologous recombination (NAHR). Considering that these LCRs share more than 96% homology, NAHR is the most likely mechanism [4,7]. Each combination can result in different chromosomal alterations, including duplications (22q11.2 duplication syndrome, OMIM #608363), inversions, supernumerary bisatellited marker chromosomes (Cat Eye syndrome, OMIM #115470), and ring chromosomes, in addition to deletions (Figure 2).
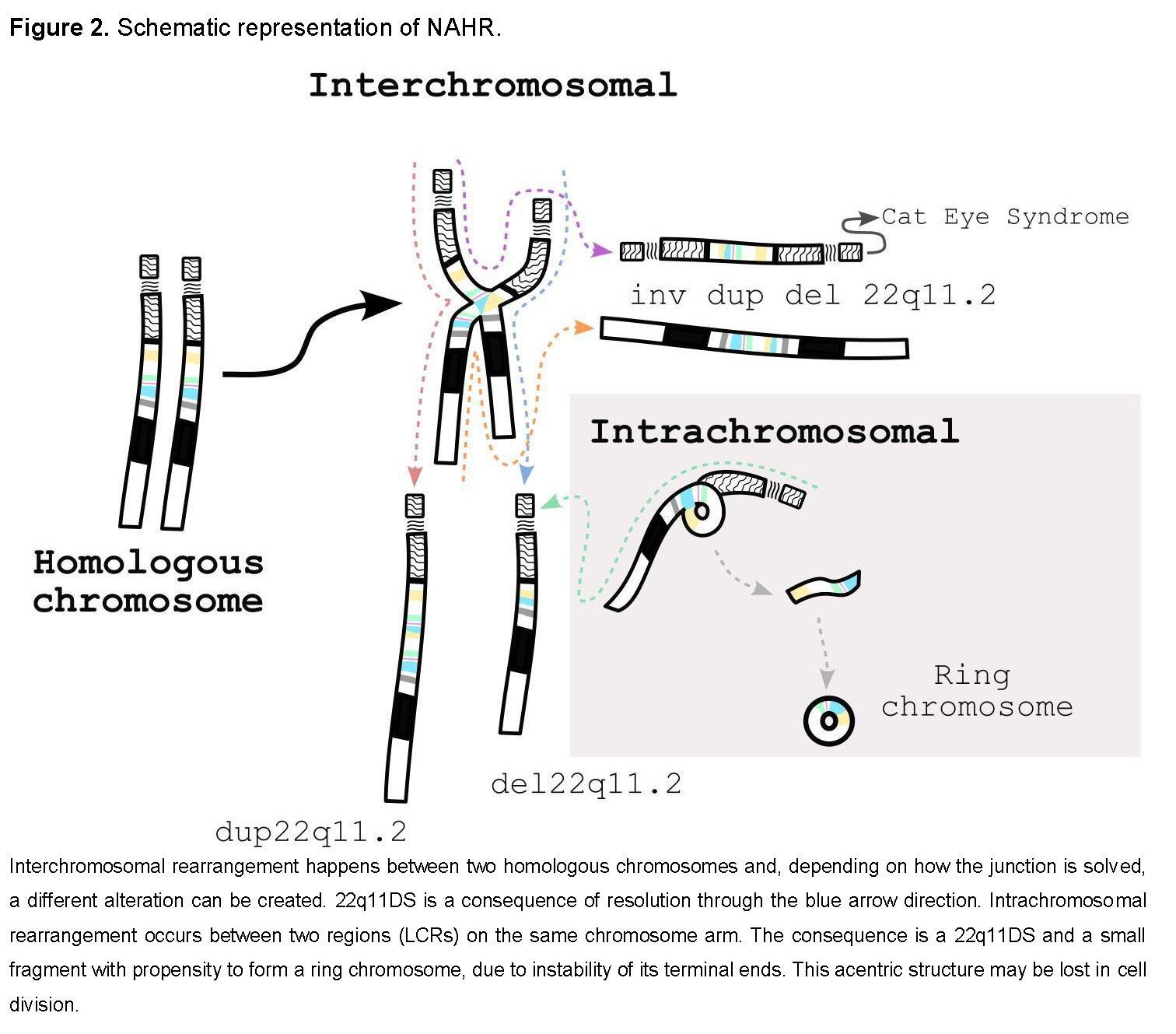
Who is causing it?
It may be inherited from parents carrying the deletion or caused by errors in gamete or embryonic divisions (Figure 3). Approximately 90–95% of cases are de novo deletions, with both sexes and all ethnic groups affected, and a slight predominance of maternal origin [8]. There is no association with parental age [2,4,7]. Even when neither parent has the deletion, there is still a slightly increased recurrence risk (RR) due to mosaicism or germline mosaicism. Germline mosaicism is suspected when two or more affected children are born to apparently unaffected parents [9]. In these cases, the RR is difficult to estimate due to multiple possible modes of segregation [10].
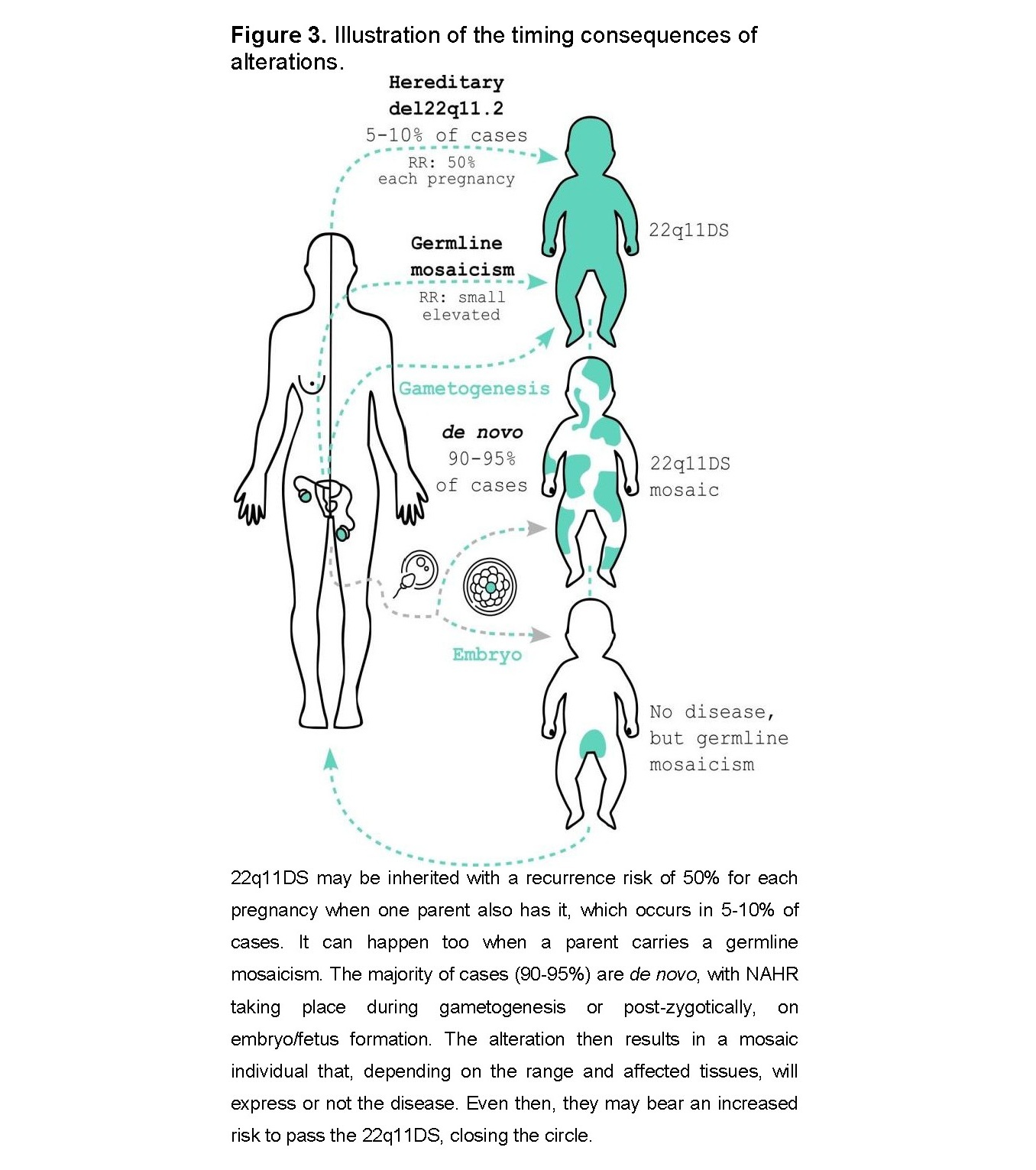
The remaining 5–10% of cases are inherited from a parent who may be unaware of their genetic condition. A full review of associated features in adulthood can be found in Boot et al., 2023 [11]. A carrier of the deletion has a 50% chance of transmitting it in each pregnancy. There appears to be strong negative selective pressure against 22q11.2 deletions mediated by the severity of the neuropsychiatric phenotype, but not necessarily by serious congenital heart disease [12].
When is it developed?
The deletion arises during meiosis or mitosis. It has been assumed that meiotic NAHR is the major mechanism in the formation of these chromosomal alterations. Interestingly, the female recombination rate in the 22q11.2 region is about 1.6 to 1.7 times higher than in males, suggesting a sex bias in meiotic processes that may explain the predominance of maternal origin [8]. A previous study supports this observation, showing that LCR22s implicated in deletions and duplications are sites of frequent meiotic recombination, mainly during oogenesis (Figure 2, dotted line) [13].
There is no evidence that the presence of one alteration predisposes to another. However, a de novo nested deletion (between LCR22A and LCR22B) has been reported together with a duplication in 70% of the cells of a girl. Her father, as well as other family members, also carries a duplication (between LCR22B and LCR22D). In this case, the proposed mechanism was mitotic rather than meiotic, involving unequal crossover during an early division after fertilization [14]. Thus, from another perspective, the de novo mutation rate could be related to ineffective repair of spontaneous DNA lesions [9], since DNA is subject to numerous damaging events daily as a result of normal cellular metabolism, and additional lesions may be generated by exogenous agents [15]. To date, no study has correlated DNA damage and repair with the occurrence of 22q11DS. Male germ cells continue to divide throughout life, allowing gradual accumulation of mutations, in contrast to female DNA recombination and repair activities, which are linked to arrested oocytes [9]. In this scenario, an increase in RR with age would be expected; however, this is not observed, although this mechanism cannot be completely excluded during gametogenesis. Nevertheless, such alterations are more likely to occur post-zygotically, during embryonic or fetal development, or even later in life [9].
Somatic NAHR is mediated by intrachromosomal exchange during mitosis and may be responsible for somatic mosaicism [16].
Where does 22q11DS take place?
The syndrome originates during gametogenesis or early embryonic development. As mentioned, the main mechanism underlying de novo 22q11DS occurs during meiosis, and therefore during gametogenesis, slightly more frequently in oocytes. Although a single meiotic event cannot result in two abnormal cell lines, a meiotic event causing duplication followed by mitotic deletion has been proposed in some cases [17]. Thus, the presence of a mosaic de novo mutation implies a post-zygotic origin, which may occur during embryonic development or even postnatally. These somatic mutations are also considered de novo because they are not detectable in the parents of affected individuals [9] (Figure 3).
Mosaicism has traditionally been considered rare and reported mainly in case reports or small case series. However, a recent study estimated a mosaicism rate of 22q11.2 deletion at 28.2%. They also observed a low level (<35%) of deleted cell lines in over 36% of cases. This finding is important for genetic counseling, as one of the parents may have low-level mosaicism in some or all tissues [16].
So, what are the consequences?
22q11DS may affect every system in the body and can be life-threatening in its most severe forms. More than 180 clinical features have been reported, associated with haploinsufficiency of approximately 90 genes identified to date. Congenital heart disease is present in 60–80% of cases and varies in severity, including a wide range of aortic arch and/or cardiac outflow tract anomalies [1,18,19]. This is also the most common condition associated with cleft palate, which may lead to velopharyngeal insufficiency and contribute to speech and language problems. Ophthalmologic abnormalities are also common. Hypoparathyroidism is diagnosed in 40–75% of cases and may present early in life as hypocalcemia leading to seizures. Immunodeficiency may occur in up to 80% of cases, with increased susceptibility to autoimmune disorders [1]. Although the severity of immunodeficiency correlates with thymic aplasia or hypoplasia, T-cell numbers may still be normal due to ectopic thymic tissue within the mediastinum [20].
Brain volume reduction (particularly in the temporal lobes and hippocampus) occurs with age, and reduction of the prefrontal cortex can lead to deficits in working memory. Intellectual disability is often present, although more than half of individuals have borderline to normal intelligence. It is estimated that 90–100% of children with 22q11.2 deletion syndrome meet criteria for a learning disability. The cognitive phenotype also includes difficult temperament and a predisposition to anxiety, depression, and attention deficits. Up to 50% of adolescents report transient psychotic episodes, while approximately 33% of adults with VCFS are diagnosed with schizophrenia [1]. The clinical manifestations in offspring may result in a more severe cognitive phenotype. The reason for this variability in clinical expressivity is not well understood.
However, several biological mechanisms appear to be involved, including sensitivity to gene dosage within the 22q11.2 region and other heritable components, such as variants in genes on the intact chromosome 22q11.2 and/or additional modifying variants outside the region, ranging from protein-coding genes to regulatory elements [7].
To date, five genes within the 22q11.2 region have been mainly implicated in the neuropsychiatric phenotype: proline dehydrogenase (PRODH), DiGeorge critical region 8 (DGCR8), catechol-O-methyltransferase (COMT), T-box 1 (TBX1), and septin 5 (SEPT5). Environmental factors may also contribute to these manifestations [21]. Apparently, mosaicism level does not correlate directly with phenotype. Low-grade mosaicism (<35% of cells with deletion) can produce the classical phenotype, while high-grade mosaicism (>65%) may result in milder presentations. This difference may be due to tissue-specific mosaicism [16].
Ultimately, deletion size and genomic location have a direct impact on clinical manifestations. Patients with proximal nested deletions (involving LCR22A) share major phenotypic features with those carrying the typical LCR22A–LCR22D deletion. In contrast, distal nested deletions (LCR22B–LCR22D and LCR22C–LCR22D) are associated with overlapping but less penetrant phenotypes and are more frequently inherited [7]. Atypical deletions, with breakpoints located between LCR22 sequences, have also been described. The prevalence of these nonstandard deletions is likely underestimated because they are more difficult to detect with current genetic testing and are associated with milder phenotypes [4].
And then, what can we do about it?
The variability of symptoms in 22q11DS makes it difficult to predict disease manifestation [1]. However, the associated health problems can significantly impact daily family life and influence reproductive decision-making. Preimplantation genetic diagnosis and prenatal diagnosis may be offered to couples at increased recurrence risk. Although there is general reluctance to terminate pregnancies affected by 22q11DS, it may be considered in regions where it is permitted [2].
Physical manifestations are often manageable. Surgical interventions can correct congenital heart defects and cleft palate, and in some cases, thymic transplantation may be required due to T-cell deficiency [1]. Regarding cognitive outcomes, environmental factors such as family dynamics, socioeconomic status, and psychological stress may influence the clinical profile, although further studies are needed to clarify these effects [21].
We do not know where science will lead us, and many questions remain unanswered: Can we prevent manifestations? What is the role of somatic mosaicism? Will a cure be found? Could targeted gene therapy addressing the underlying cause provide a cure? Or will we be able to modify the disease course? Could this condition eventually be eradicated?
The possibility of eradicating this disease is complex because the genomic architecture predisposes to recurrent 22q11DS events. Nevertheless, increasing our understanding of the biological functions of genes within the 22q11.2 region and how environmental factors influence their expression may help clarify the heterogeneity of this condition [21] and guide future therapeutic strategies. Continuous advances in knowledge, appropriate public health policies, and precise diagnosis are essential to unravel the complexity of this and other genomic conditions.
ABREVIATIONS
22q11DS: 22q11.2 deletion/DiGeorge Syndrome; COMT: Catechol-o-methyltransferase; DGCR8: DiGeorge Critical Region 8; LCRs: Low copy repeats; NAHR: Nonallelic Homologous Recombination; PRODH: Proline dehydrogenase; RR: Recurrence Risk; SEPT5: Septin 5; TBX1: T-box 1.