
INTRODUCTION
Diploid/triploid mosaicism is a rare genetic alteration characterized by the coexistence of cell lines with different chromosome numbers in the same individual. Although the exact prevalence is unknown due to its rarity, understanding its clinical and genetic impact is essential. This disorder presents with a wide spectrum of clinical phenotypes, ranging from minor anomalies to severe conditions involving overgrowth and body asymmetry (Happle, 1987; Ledbetter et al., 1992).
Hemihypertrophy, one of the manifestations of diploid/triploid mosaicism, is a condition in which one part of the body is noticeably larger than the other, and it is associated with a variety of genetic syndromes (Hoyme et al., 1998). Early diagnosis and monitoring are crucial due to the increased risk of certain types of cancer in patients with hemihypertrophy (Clericuzio and Martin, 2009).
Cases of diploid/triploid mosaicism often present diagnostic challenges, particularly in the prenatal context. The presence of a molar placenta, as observed in this case, can be an early indicator of this condition (Vogel et al., 2010). Accurate identification of mosaicism is essential for clinical management and genetic counseling, as it has significant implications for treatment and prognosis (Kalousek et al., 1993).
This clinical case highlights the importance of detailed genetic evaluation in the presence of prenatal and postnatal developmental anomalies. Through a multidisciplinary approach integrating genetics, orthopedics, radiology, and pediatrics, an accurate diagnosis and optimal management plan can be achieved for these complex patients (Biesecker and Spinner, 2013).
METHODOLOGY
The inclusion criteria involved:
- Patient with a phenotype compatible with diploid/triploid mosaicism.
- Evidence of body asymmetry and hemihypertrophy.
- Prenatal findings suggestive of chromosomal abnormalities.
- Confirmation of chromosomal abnormalities through genetic testing.
The exclusion criteria involved:
- Cases with a confirmed alternative etiology for body asymmetry.
- Patients with clinical findings not compatible with diploid/triploid mosaicism.
- Lack of informed consent.
Description of Tests Performed:
- Whole Exome Sequencing (WES):
- Enzymatic fragmentation of genomic DNA.
- Target region enrichment using capture probes.
- Sequencing on an Illumina platform with >98% coverage of the human exome.
- Bioinformatic analysis for the identification of pathogenic variants.
- SNP Microarrays (Array-CGH):
- Use of the Infinium™ Global Diversity Array with Cytogenetics (Illumina) kit.
- Evaluation of CNVs and aneuploidies in the genome.
- Analysis using NxClinical software (BioDiscovery).
- Microsatellite (STR) Analysis:
- Evaluation of 15 autosomal STR markers and amelogenin.
- Use of the PowerPlex 16HS multiplex kit (Promega).
- Confirmation of the parental origin of the triploid chromosomal complement.
- Karyotyping in Peripheral Blood and Affected Tissue:
- Lymphocyte culture for cytogenetic analysis.
- Evaluation of fibroblasts from affected tissue to detect mosaicism.
- Confirmation of 46,XX in peripheral blood and 69,XXX in affected tissue.
- Imaging Tests:
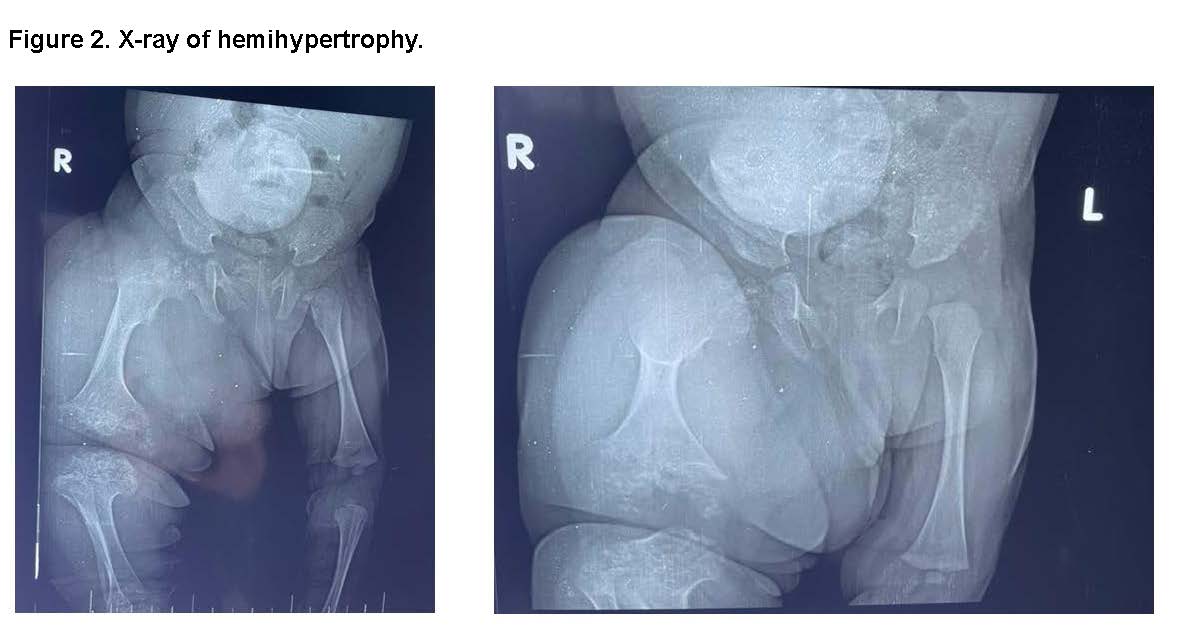
- X-ray of the right leg: Bone dysplasia and overgrowth.
- Soft tissue ultrasound: Increased tissue volume in the right lower limb.
- Arterial and venous Doppler: No vascular abnormalities.
- Echocardiogram: Patent foramen ovale and minimal pericardial effusion.
- Abdominal ultrasound: Giant umbilical hernia and lipohypertrophy.
Complementary Clinical Evaluations:
- Ophthalmologic and auditory evaluation: Normal findings.
- Anthropometric measurement and detailed physical examination.
Bioethical Considerations: We declare having obtained informed consent from the patient for this publication, ensuring their understanding of the objectives, implications, and scope of the information disclosure.
This publication adheres to the ethical principles established in the Declaration of Helsinki of the World Medical Association, which governs research involving human subjects, ensuring respect for the autonomy, confidentiality, and well-being of the participants.
Sequencing methodology
In whole exome sequencing, genomic DNA was enzymatically fragmented and target regions were enriched using DNA capture probes. These regions include approximately 41 Mb of the human coding exome (targeting >98% of the RefSeq coding regions of the human genome build GRCh37/hg19), as well as the mitochondrial genome. The generated library was sequenced on an Illumina platform to achieve a read depth of at least 20x for >98% of the targeted bases. An in-house bioinformatics pipeline was applied, which includes alignment of the reads to the GRCh37/hg19 genome assembly and the revised Cambridge Reference Sequence (rCRS) of the human mitochondrial DNA (NC_012920), variant calling, annotation, and comprehensive variant filtering. All variants with a minor allele frequency (MAF) below 1% in the gnomAD database and disease-causing variants reported in HGMD®, ClinVar, or CentoMD® were evaluated. The investigation of relevant variants focused on coding exons and ±10 intronic nucleotides flanking genes with clear gene-phenotype associations (based on OMIM® information). All potential inheritance patterns were considered. Additionally, family history and the clinical information provided were used to evaluate the identified variants with respect to their pathogenicity and disease causality. Strict quality criteria and validation processes were established for variants detected by NGS. Variants with low sequencing quality and/or unclear zygosity were confirmed using orthogonal methods. Consequently, a specificity >99.9% is guaranteed for all reported variants.
For array analysis, the Infinium™ Global Diversity Array with Cytogenetics (Illumina) kit was used. This kit contains ~1.8 million SNP markers distributed across the entire genome, particularly targeting more than 4800 disease-associated genes with 99.9% exonic coverage, enabling the detection of copy number variations (CNVs). Genomic DNA was amplified, fragmented, and hybridized to the array according to the manufacturer's instructions. The results were analyzed using NxClinical software (BioDiscovery). In the case of microsatellites, 15 autosomal STR markers and amelogenin were analyzed using the PowerPlex 16HS multiplex kit (Promega Corporation, Madison, USA).
RESULTS
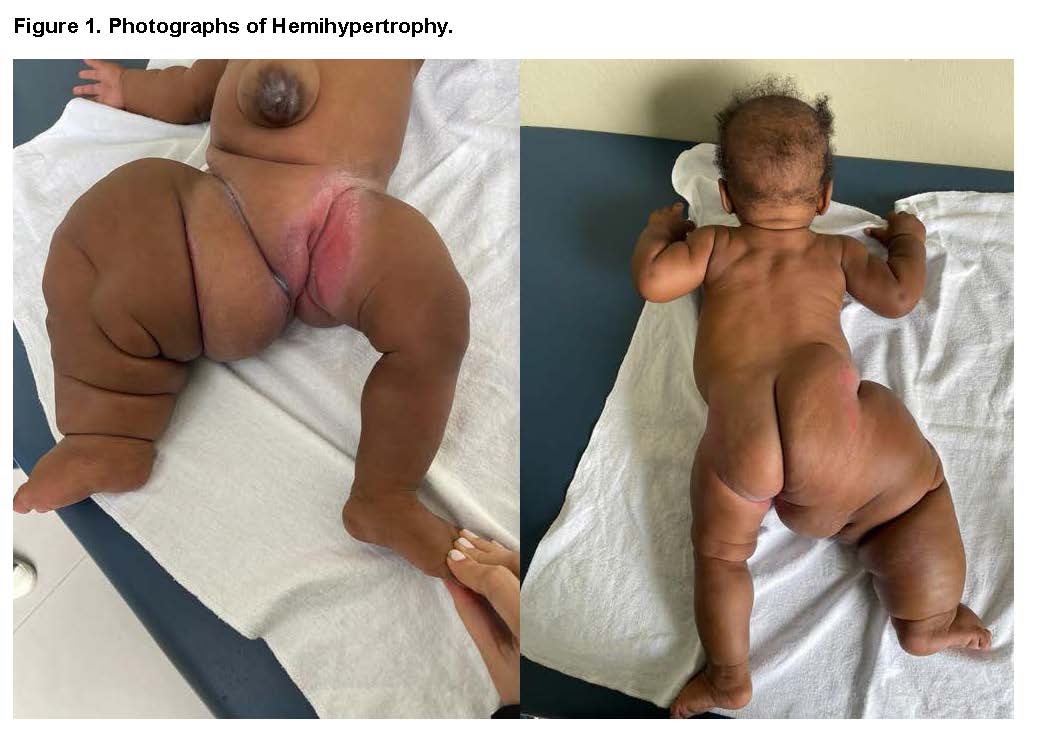
The patient is a one-year-old female, born to non-consanguineous parents—a 20-year-old mother (G2P2C0A0) and a 26-year-old father. Prenatal follow-up included regular check-ups, with the identification at 31 weeks of gestation of a placental abnormality characterized by molarity or mesenchymal dysplasia, detected via ultrasound. This condition coexisted with a live fetus presenting with right leg hypertrophy and skeletal dysplasia, as well as mild polyhydramnios. At 36 weeks, a discrepancy in fetal leg size was observed. The patient was born at term via vaginal delivery, weighing 6 pounds, and was discharged after five days of uneventful observation.
Placental biopsy revealed a mature monochorionic monoamniotic placenta with signs of focal anoxia, edema, hemorrhage, and focal acute chorioamnionitis. Additionally, hydropic changes were observed in the villi, with cistern formation and mild trophoblastic hyperplasia, suggesting a partial incomplete molar component and the possibility of cytogenetic anomalies and placental mesenchymal dysplasia.
The patient exhibited delayed motor development and has a history of cow's milk protein allergy. She has been hospitalized twice due to urinary tract infections. There is no reported family history of medical relevance.
Diagnostic Test
Right Leg X-Ray: Revealed shortening of the diaphysis and widening of the epiphysis, suggestive of skeletal dysplasia of the right lower limb.
Soft Tissue Ultrasound: Detected increased soft tissue in the right lower limb.
Arterial and Venous Doppler: No evidence of vascular pathology in the limb.
Echocardiogram: Identified a patent foramen ovale and a minimal pericardial effusion.
Ophthalmologic and Hearing Evaluations: Results were normal.
Abdominal Ultrasound: Revealed an abdominal wall defect and a giant umbilical hernia.
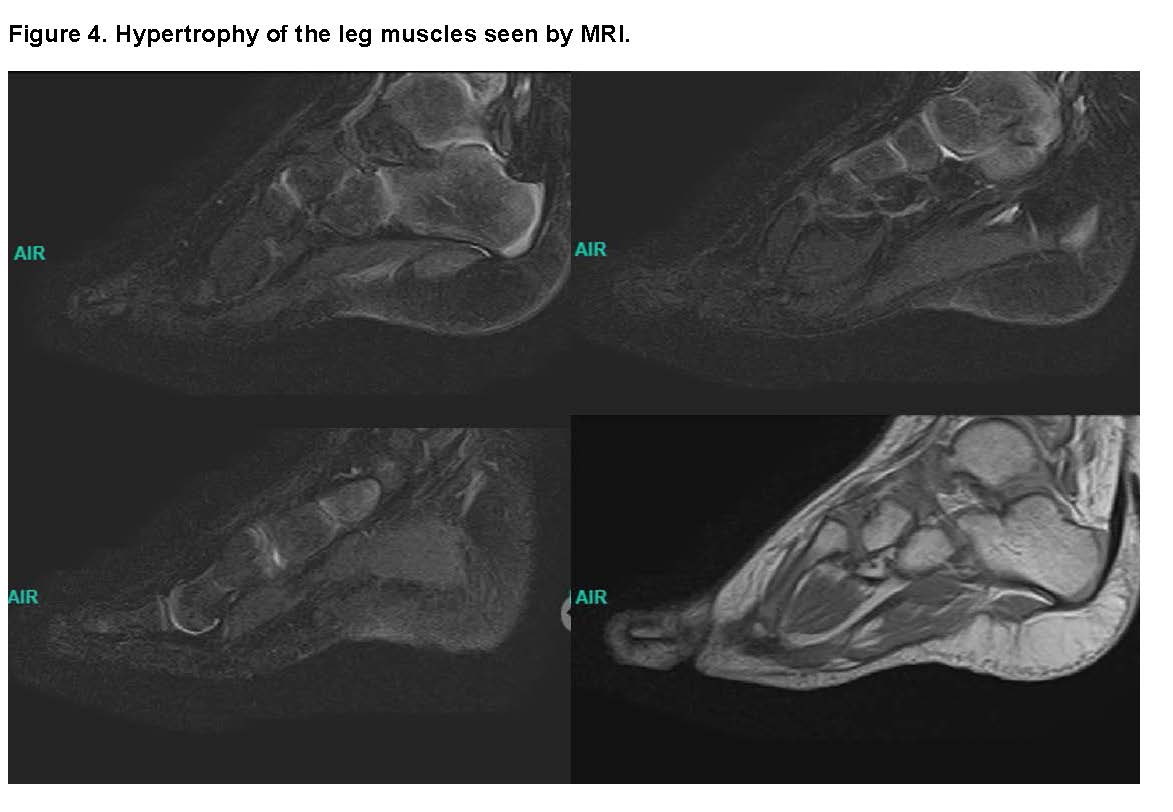
MRI of the Right Leg: showed an Hyperplasia of the tibia with absence of the growth plate and morphological distortion with widening of the tibial plateau, an Hypertrophy of the lateral malleolus with an abnormal appearance and a Hypertrophy of leg muscles except for the gastrocnemius, which shows atrophy and fatty replacement.
MRI of the Right Thigh: Femoral head dysplasia with hypoplasia of the femoral diaphysis. Probable acetabular insufficiency based on morphological data. Muscle fibrosis with fatty replacement of quadriceps muscle fibers. Generalized muscle hypertrophy.
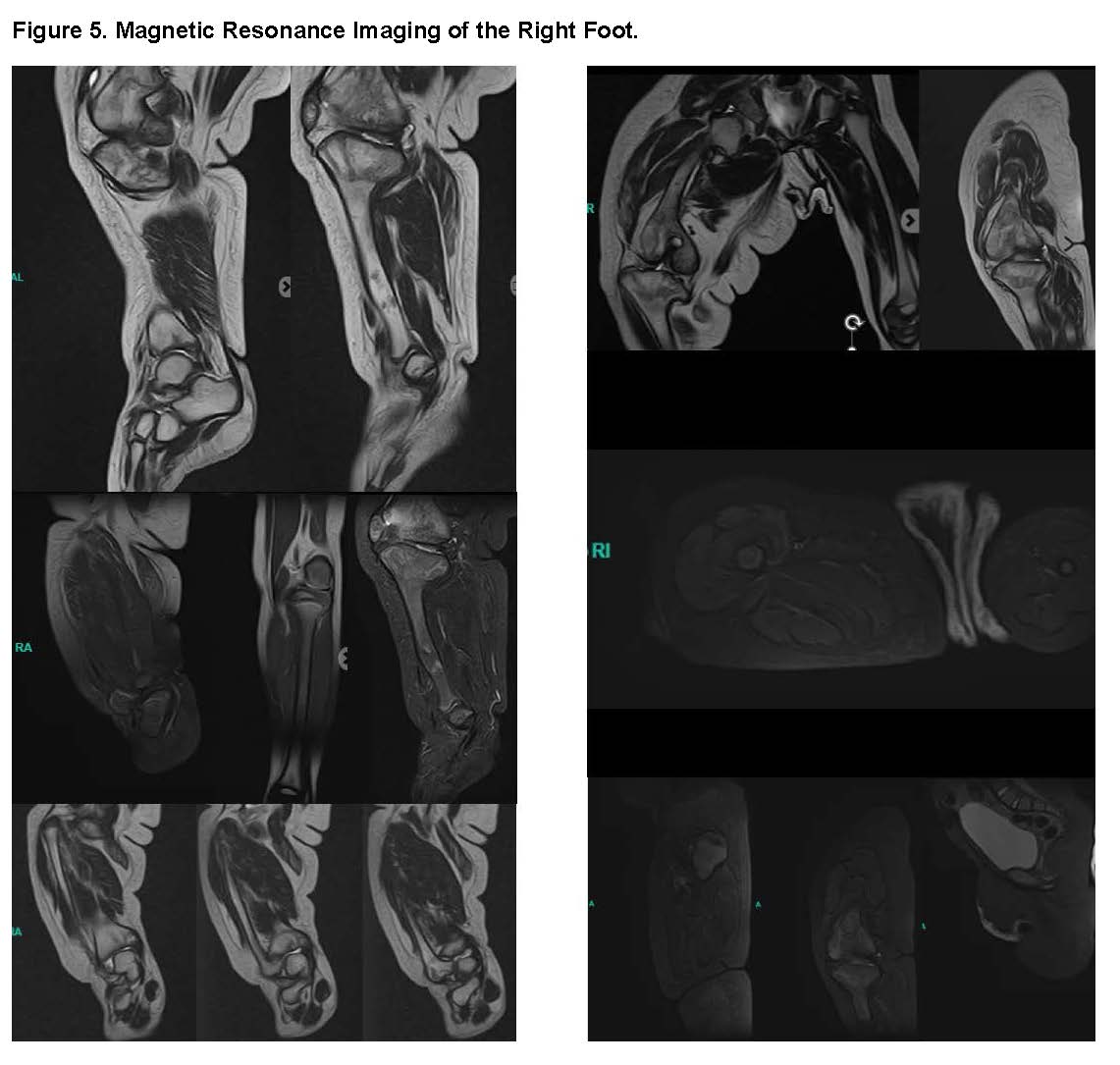
MRI of the Right Foot: The MRI showed a Calcaneal-talar divergence and Shortening of the Achilles tendon.
Brain MRI: The brain MRI showed no evidence of structural abnormalities, lesions, masses, signs of infection or inflammation, or vascular abnormalities. The morphology and signal of intracranial structures were normal, including white and gray matter, ventricles, and subarachnoid spaces. No signs of cerebral edema or midline shift were observed. The study is consistent with a neurological exam without pathological findings.
The Anthropometric Measurements are described below:
- Weight: 9 kg (50th percentile)
- Height: 85 cm (>97th percentile)
- Head circumference: 44.8 cm (25th percentile)
During the physical examination, cranial asymmetry, high hairline, frontal bossing, epicanthus, telecanthus, flattened nasal bridge, and other dysmorphic features were observed. The right leg is significantly enlarged with altered skin folds, suggesting an overgrowth syndrome with body asymmetry.
During genetic evaluation of the affected tissue, DNA analysis (right leg with hemihypertrophy) showed a triploid chromosomal complement: arr(X,1-22)x3. Microsatellite marker fragment analysis revealed a diallelic trisomic pattern (2:1) for 11 informative STRs. NGS data and SNP profile from microarray analysis (used as internal control) were consistent with a triploid genome (69,XXX). The chromosomal anomaly is classified as pathogenic based on ACMG guidelines.
In the peripheral blood DNA analysis, no clinically relevant DNA sequence variants, copy number variations, or other findings associated with the reported phenotype were detected in the peripheral blood DNA sample. Exome sequencing showed a normal female diploid chromosomal complement, confirmed by chromosomal microarray and consistent with previous patient karyotyping: arr(X,1-22)x2 htz.
Karyotype Results: Peripheral blood testing gave out a normal female karyotype of 46XX. However, the right leg has an abnormal karyotype of a triploidy of 69 XXY.


DISCUSSION
This case of diploid/triploid mosaicism represents the first documented report in the Dominican Republic, highlighting the importance of its detailed description and analysis. The patient presents a phenotype characterized by hemihypertrophy, overgrowth, and skeletal abnormalities—findings that align with those reported in previous studies on this condition (Happle, 1987; Ledbetter et al., 1992; Hoyme et al., 1998). However, the combination of these features with prenatal history of placental dysplasia and vascular anomalies reinforces the phenotypic heterogeneity of diploid/triploid mosaicism.
The genetic findings in this patient emphasize the importance of analyzing samples from affected tissue, as the peripheral blood karyotype was normal (46,XX), whereas triploidy (69,XXX) was confirmed in the tissue from the hemihypertrophic leg. This finding is consistent with previous reports where peripheral blood does not always reflect the presence of mosaicism (Van De Laar et al., 2002; Boonen et al., 2011). Additionally, the microsatellite study confirmed a diallelic trisomic pattern (2:1), indicating that the extra set of chromosomes is of maternal origin, in line with the hypothesis of second polar body inclusion during embryogenesis (Brems et al., 2003).
A comparative analysis with the medical literature reveals several phenotypic similarities and differences. In previous studies, diploid/triploid mosaicism has been associated with growth delay, hypotonia, facial anomalies, and skeletal dysplasias (Graham et al., 1981; Rittinger et al., 2008). In our case, similar anomalies were identified, although the patient presents a more pronounced overgrowth phenotype, which could be related to the tissue-specific distribution of triploid cells. This aligns with studies suggesting that phenotypic expression depends on the percentage of triploid cells in specific tissues (Oktem et al., 2007).
From a clinical standpoint, long-term follow-up is essential. An increased risk of neoplasia has been described in patients with hemihypertrophy, particularly in those with underlying genetic alterations (Clericuzio & Martin, 2009). In this context, the patient requires periodic monitoring through abdominal ultrasound and additional tests for the early detection of associated tumors. The literature supports this approach, emphasizing the need for regular evaluations and multidisciplinary follow-up (Biesecker & Spinner, 2013).
From a bioethical perspective, managing patients with diploid/triploid mosaicism presents challenges in genetic counseling, as its clinical expression can be highly variable. In our case, detailed information was provided to the family regarding the uncertain prognosis and the need for genetic studies on affected tissue samples for more accurate evaluation. This is consistent with reports that stress the importance of genetic counseling in rare conditions and the need for individualized management (Kalousek et al., 1993; Vogel et al., 2010).
This case significantly contributes to the understanding of diploid/triploid mosaicism, emphasizing the importance of genetic analysis in affected tissue for accurate characterization. The observed phenotype confirms the clinical variability of the condition and reinforces the need for a multidisciplinary approach in the management of these patients. Periodic surveillance and genetic follow-up remain essential aspects to ensure optimal and personalized care.
Acknowledgments
To Jessie PEPÉN BODRÉ