
INTRODUCTION
The MTHFR gene (Methylenetetrahydrofolate Reductase), located on chromosome 1p36.22, plays an essential role in folate metabolism and the methylation of homocysteine to methionine. This metabolic pathway is critical for the synthesis of DNA, RNA, and proteins, and its dysfunction can lead to significant clinical implications [4,5]. Mutations in MTHFR, particularly the C677T and A1298C polymorphisms, have been linked to multiple pathologies, including neural tube defects, psychiatric disorders such as schizophrenia, thromboembolisms, and homocystinuria due to MTHFR deficiency [1,2,5]. The latter is inherited in an autosomal recessive manner and is characterized by the accumulation of homocysteine in blood and urine, leading to multisystemic toxicity [2,3].
The clinical spectrum of homocystinuria includes neurological alterations (cognitive delay, seizures), ocular (lens dislocation), skeletal (tall stature, Marfan-like deformities), and vascular (increased thrombotic risk) [3]. The evidence compiled in the OMIM database integrates genetic association studies, meta-analyses, clinical studies, experimental studies, and scientific letters, reflecting the complexity of the impact of MTHFR at the molecular and population levels [1].
Early identification of pathogenic variants and their correlation with biomarkers such as homocysteine allows for a preventive and personalized therapeutic approach [3,4]. This compendium underscores the importance of genetic screening and dietary intervention (such as folate supplementation) in vulnerable populations [3,6]. Thus, MTHFR is established as a key genetic marker in personalized medicine and in understanding complex diseases with a metabolic and epigenetic basis [4,5,7].
The MTHFR gene (OMIM 607093) encodes a key enzyme in folate and homocysteine metabolism, essential processes for DNA methylation and methionine synthesis. Genetic variations in this gene, such as the C677T and A1298C polymorphisms, have been associated with multiple pathologies, including neural tube defects, thrombosis, and neuropsychiatric disorders [1,2,4]. This research explores the clinical implications of these mutations and the relevance of MTHFR as a genetic marker in personalized medicine [5,6].
METHODOLOGY
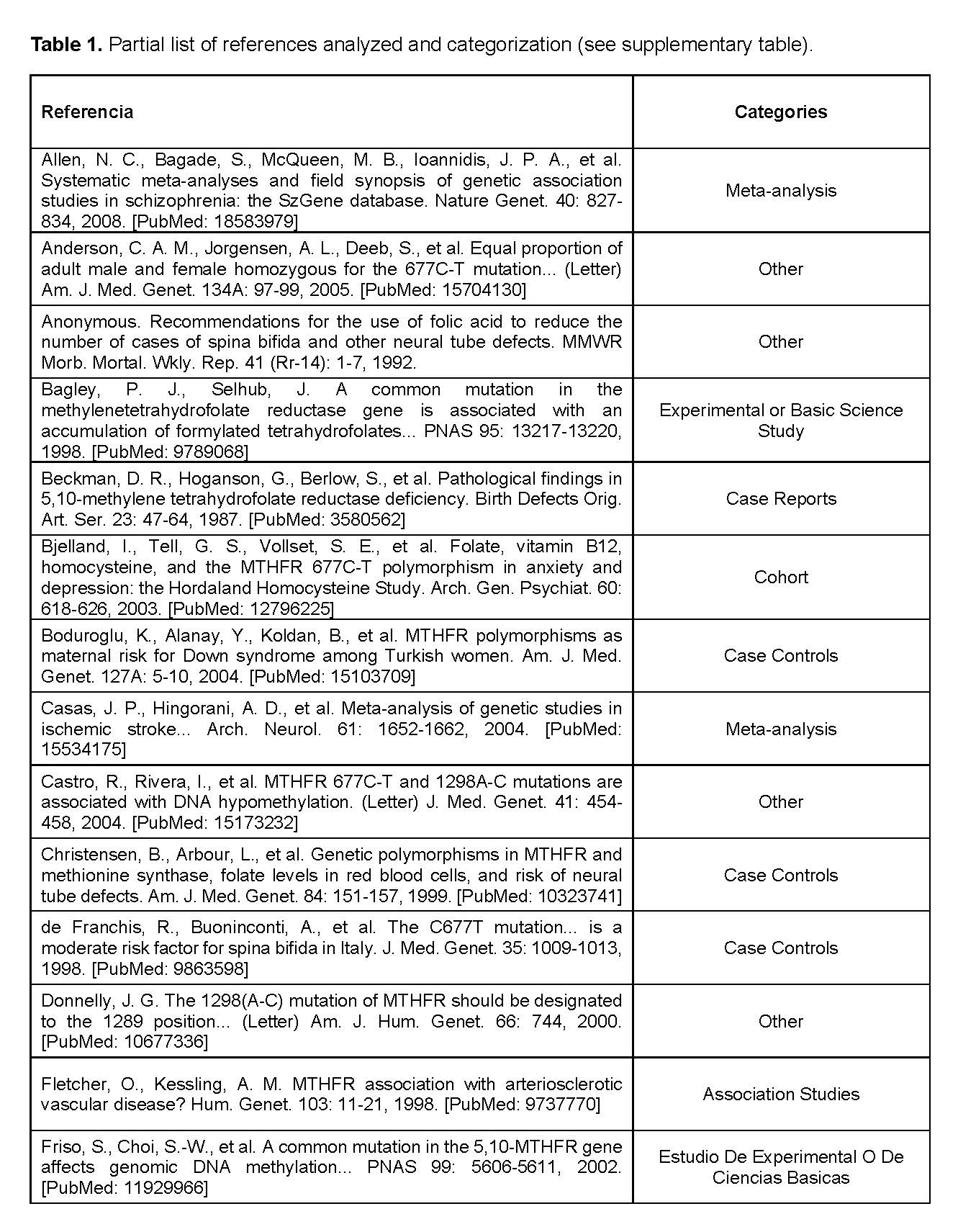
For the completion of this work, our group adopted a collaborative and efficient organization. From the early stages, we conducted planning meetings to define the content and distribute tasks based on the individual strengths of each member. Specific responsibilities were assigned: some members focused on information gathering, others on writing the text, and others on designing the presentation. All the information used was obtained from the OMIM site, which ensured the truthfulness and solidity of the content [1]. Throughout the process, we maintained constant communication to ensure the coherence and proper structuring of all sections. This collaborative approach allowed us to develop a comprehensive, rigorous, and well-founded project. Each article was assigned to one of 8 possible categories depending on the type of study. The categories were: association studies, case-control, experimental, meta-analysis, case report, negative association, transmission study, cohort, and others.
RESULTS
The genetic function of the MTHFR gene is unknown; this gene focuses on the production of the enzyme methylenetetrahydrofolate reductase (EC 1.5.1.20). This enzyme catalyzes the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate. 5-methyltetrahydrofolate is an essential substrate for the remethylation of homocysteine to methionine [1,2]. The MTHFR gene is located in the 1p36.22 region of chromosome 1 and is composed of 11 exons in humans. Several polymorphisms have been identified in this gene, which can affect the enzyme's activity [1,5]. In the case of homocystinuria, it is due to MTHFR deficiency, which is a disease where there is a deficiency of the methylenetetrahydrofolate reductase (MTHFR) enzyme. This enzymatic deficiency is due to mutations in the MTHFR gene [2]. Elevated levels of homocysteine in blood and urine are characteristic of this condition [3]. The inheritance pattern for homocystinuria due to MTHFR deficiency described in the sources is autosomal recessive [1,2].
Diseases associated with MTHFR OMIM 607093
- Neural tube defects [1,2]
- Schizophrenia [2,6]
- Thromboembolism [3,5]
- Homocystinuria due to MTHFR deficiency [2,3]
The evidence on haploinsufficiency and triplosensitivity in relation to the MTHFR gene is limited and, to date, a conclusive correlation has not been established in recognized clinical databases such as ClinGen [1]. Haploinsufficiency refers to the inability of a single functional copy of the gene to maintain normal function, while triplosensitivity implies a pathological phenotype due to the presence of an additional copy of the gene. In the case of MTHFR, the main variants of interest (such as C677T and A1298C) generate quantitative enzymatic changes, but clinical phenotypes attributable to a single missing copy or duplication of the locus have not been consistently described [1,7]. The clinical effects observed are usually related to homozygous or compound heterozygous combinations in the context of functional polymorphisms, rather than with complete deletions or duplications of the gene. Therefore, according to the current evidence, there is insufficient support to demonstrate haploinsufficiency or triplosensitivity as primary disease mechanisms for MTHFR, indicating the need for more structural and functional studies to define its possible impact in these genomic contexts [1,7].
Homocystinuria is a genetic disease caused by mutations in the CBS gene, which encodes the enzyme cystathionine beta-synthase, essential for the metabolism of the amino acid methionine. When this gene is altered, the enzyme does not function correctly, preventing the conversion of homocysteine into cystathionine. As a result, homocysteine accumulates in the blood and urine, producing toxic effects on various organs [3]. This accumulation causes neurological disorders, ocular alterations such as lens dislocation, skeletal malformations, and a high risk of thrombosis [3]. Homocystinuria is a clear example of how a single genetic mutation can generate a multisystem disorder with serious consequences if not detected and treated in time [3]. When there is a mutation in this gene, enzymatic activity is reduced or completely lost. Consequently, homocysteine is not adequately metabolized and accumulates in blood, urine, and tissues, causing toxic effects through various mechanisms, including oxidative stress, endothelial damage, and alteration of sulfur metabolism [3]. This accumulation severely affects multiple systems of the body. Clinically, homocystinuria manifests through neurological alterations (mental retardation, seizures, behavioral changes), ocular defects (lens dislocation, severe myopia), vascular problems (increased risk of venous or arterial thrombosis), and skeletal alterations (tall stature, scoliosis, deformities similar to Marfan syndrome) [3]. The variety of clinical manifestations, which can appear in early stages or progress insidiously, highlights the importance of early diagnosis [3].
In our evaluation of the references of this gene in OMIM, we were able to classify a total of 113 articles. The vast majority were clinical association studies, followed by non-clinical experimental studies. In frequency followed 14 case-controls, 10 meta-analyses and 10 case reports.
It is important to note that only 2 cohort studies and 2 transmission studies were identified. The Other Studies group represented a total of 24 cases (see Table 1 and Graph 1).
DISCUSSION
The findings presented reinforce the clinical relevance of the MTHFR gene as a key component in folate and homocysteine metabolism, fundamental processes for neurological, vascular, and reproductive health. The high frequency of genetic association and case-control studies in the reviewed literature indicates a sustained interest in understanding the most common polymorphic variants, such as C677T and A1298C, in various populations and diseases. Despite the abundant evidence, some controversy persists regarding the clinical interpretation of these variants, especially in contexts such as mental health, fertility, and response to nutritional treatments.
It is noteworthy that multiple studies not only confirm significant associations but also point out negative or inconclusive results, suggesting the influence of epigenetic, environmental, and gene interaction factors. Likewise, the limited evidence of haploinsufficiency and triplosensitivity according to ClinGen highlights the need for more robust functional studies to clarify the impact of structural alterations in this gene.
In our evaluation of the references related to the MTHFR gene in the OMIM database, we were able to classify a total of 113 articles, observing a clear predominance of clinical association studies (n = 32), followed by experimental studies (n = 14) and case-controls (n = 14). This distribution suggests a priority focus on observational clinical and genetic research aimed at establishing correlations between MTHFR gene variants and various pathologies.
The presence of 4 letters to the editor and 4 negative association studies is noteworthy, indicating that there is also a critical and contradictory body of evidence that deserves consideration when interpreting the findings. Particularly notable is the low representation of cohort studies (n = 2) and transmission studies (n = 2), reflecting a limitation in broad longitudinal and molecular approaches. These findings highlight the need to diversify the methodological design in future research to strengthen the comprehensive understanding of the clinical and functional impact of the MTHFR gene (See Table 1 and Graph 1).
CONCLUSION
The MTHFR gene represents a relevant genetic marker in personalized medicine because of its role in folate and homocysteine metabolism. In our review of 113 references in OMIM, we observed a predominance of association studies (n = 32), followed by genetic association studies and meta-analyses, evidencing an observational approach in the literature. The low presence of cohort and genomic studies indicates the need to diversify methodological designs. These findings underscore the importance of interpreting genetic results with caution and promoting more robust and varied research, and allow us to evaluate the clinical evidence of this gene.
About the authors
EM, CF, RR, SR, LF are medical students at the Interamerican University of Panama. JMR is a professor of genetics and immunology at the Interamerican University of Panama, and Clinical Specialist in Qu bec.
Author Contributions
EM,CF, RR, SR, LF curation, analysis and writing. JMR conceptualization, analysis and writting.