
CASE REPORT
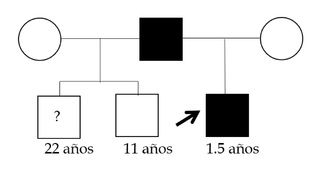
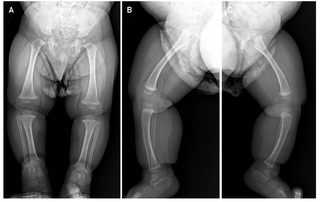
This is a 2-year-old male infant, first child of non-consanguineous parents, with no relevant history of pregnancy and delivery at term and without complications, who is referred to the Genetics consultation by his pediatrician for severe short stature (- 5DS), and with an ongoing diagnostic suspicion process for skeletal dysplasia. The patient has two paternal half-siblings (Figure 1). On physical examination the patient had mild dysmorphologic findings including midface hypoplasia, flat nasal bridge, relative macrocephaly, frontal bossing, brachydactyly and lordosis (Figure 2). Radiographic evaluations at 11 months of the entire skeleton revealed as the only notable finding cup widening of both femurs with discrete longitudinal shortening of both tibias with anteroposterior curvature suggestive of bone dysplasia (Figure 3), and with a bone age study, which revealed an advanced bone age for the age of 1 year and 3 months (4 months). The patient received early stimulation due to the detection of a slight delay in psychomotor development with hypertonia, and at one year of age he was meeting all the developmental milestones. Molecular genetic testing was performed in the evaluation process.

Molecular Tests
A Whole Genome Sequencing (WGS) study with Multiomic Analysis (Centogenome ® MOx 2.0) was performed at Centogene, involving also the transcriptomic study. Genomic DNA was enzymatically fragmented and labeled with Illumina-compatible adapter sequences. Libraries were sequenced from both ends (paired-end) on an Illumina platform to generate an average coverage depth of ~ 30x. A bioinformatics process based on Illumina's DRAGEN process as well as CENTOGENE's in-house bioinformatics process was applied. Reads were aligned to the Genome Reference Consortium Human Build 37 (GRCh37/hg19) genome assembly, as well as to the revised Cambridge Reference Sequence (rCRS) of human mitochondrial DNA (NC_012920). Sequence variants (SNVs/indels) and copy number variants (CNVs) were called using the DRAGEN algorithm, Manta and proper. All variants with a minority allele frequency (MAF) of less than 1% in the gnomAD database, and disease-causing variants reported in HGMD®, in ClinVar or in the CENTOGENE Biodatabank were evaluated.
Although the evaluation focused on coding exons and flanking intronic regions, the entire gene was interrogated for candidate variants with a plausible association with phenotype. All potential patterns of mode of inheritance were considered. In addition, family history and clinical information provided were used to evaluate the identified variants with respect to their pathogenicity and disease causation. Variants are classified into five classes (pathogenic, probably pathogenic, VUS, probably benign, and benign) following ACMG guidelines for variant classification in addition to ClinGen recommendations. All relevant variants related to the patient's phenotype were reported. RNA was isolated from CentoCard®. Messenger RNA (mRNA) was enriched, fragmented and transcribed into complementary DNA (cDNA). This was followed by ligation of adapters and amplification of libraries, which were pairwise sequenced on an Illumina platform. Sequencing data were processed using a proprietary bioinformatics pipeline. Sequencing reads were evaluated using quality control tools to ensure data integrity and identify potential problems (low quality reads, adapter contamination, etc.). The reads were then aligned against the GRCh37/hg19 reference genome using STAR, which allows accurate determination of splicing events. Quality criteria were applied to accept or reject the sequencing data. Accepted data were analyzed and used as further evidence to assess the effect of the variant on the splicing process.
RESULTS
A variant was detected in the ACAN gene, consisting of c.2591del p.(Pro864Leufs*81) in heterozygous state, which causes a change in the reading frame starting at codon 864. The new reading frame ends in a stop codon 80 positions forward. It is classified as probably pathogenic (class 2) according to CENTOGENE and ACMG recommendations (Table 1). Pathogenic variants in the ACAN gene are associated with Kimberley-type spondyloepiphyseal dysplasia (SEDK) and short stature and advanced bone age, with or without osteoarthritis and/or early onset osteochondritis dissecans (SSOAOD).

Obtained these results and in discussion with the patient's parents, previously uncommented data emerged regarding the patient's father and one of the patient's half-siblings. The father had herniated disc surgery at the age of 43 and the older half-brother had a knee condition with rupture and degenerative changes in his meniscus. Based on these data and the indication of the diagnostic process, we proceeded to perform the ACAN gene sequencing study in the parents, detecting the same mutation in the proband's father. At the time of writing, it had not been possible to investigate the patient's half-siblings.
DISCUSION
The integrity of the development and function of the supporting elements of the organism, among which are those of the skeletal system and the extracellular matrix, depends in turn on the molecular integrity of its components. Genetic alterations in these components can produce anomalies in the structure and size of individuals.
Among these molecular structural elements are glycosaminoglycans and proteoglycans. Glycosaminoglycans (GAGs) are polysaccharides of mesodermal origin, characteristic of animal tissues and consist of heteropolysaccharides made up of large unbranched complex carbohydrates composed of repeating disaccharide units. Proteoglycans (PG), on the other hand, have a structure formed by glycosaminoglycans attached to a central protein skeleton or "core" [1].
PGs are highly variable, ubiquitous molecules with multiple roles in the organism apart from the structural role already mentioned, including cell division, adhesion, extension, migration, chemoattraction, axon guidance, extracellular matrix assembly, lipoprotein uptake, proteolysis and virus entry into cells, and interact with many molecules such as growth factors, growth factor binding proteins, extracellular proteases, protease inhibitors, protease inhibitors, extracellular matrix molecules, and are able to control gradients and availability of cytokines and chemokines, growth factor binding proteins, extracellular proteases, protease inhibitors, extracellular matrix molecules and are able to control gradients and availability of cytokines, chemokines and morphogens among other molecules and in diverse cellular circumstances and environments, such as those involved in homeostasis, embryonic development, repair and in the mechanism of diseases such as cancer [1,2]. Despite this great diversity of locations and roles, they comprise the relatively small number of 50 genes related to the central protein skeleton [3]. One of several ways to classify PGs is to divide them into families based on their predominant localization, resulting in four main classes (intracellular, cell surface, pericellular and extracellular) [4].
Genetic alterations in PGs have been associated with several conditions, apart from various types of cancer, including Alzheimer's disease, amyloid-related disorders, implications in infectious processes such as SARS-CoV-2, atherosclerosis and Ehlers-Danlos syndrome, among others [3,5].
Closely related to Ehlers-Danlos syndrome and its very diverse manifestations are those related to other PG defects in the extracellular matrix with a markedly structural effect and that produce skeletal alterations. Among the PG families with extracellular predominance, there is the Hyalectan/Lectican family that includes four genes: ACAN, VCAN, NCAN and BCAN, corresponding to the PGs aggrecan, versican, neurocan and brevican, respectively [4].
Among these, the ACAN gene mapped at 15q26.1, produces Aggrecan, which is a 250 kDa protein to which about 100 chains of chondroitin sulfate and about 30 chains of keratan sulfate are attached. It has a predominantly structural function, as it constitutes the main component of the vertebrate cartilage matrix, particularly in the cartilage or growth plate (facilitating endochondral bone formation) and in the articular cartilage, providing, thanks to its hydrated gel characteristics, the necessary conditions to fulfill the weight-bearing function performed by the joints [6]. and S Dateki summarizes the molecular characteristics of Aggrecan and the findings of at least 25 mutations in the ACAN gene associated with syndromic and non-syndromic short stature [7], which together give rise to a set of conditions known as Aggrecanopathies, and which mainly include Spondyloepiphyseal Dysplasia Kimberley type (SEDK), Familial Osteochondritis Dissecans, Macrocephaly with Multiple Epiphyseal Dysplasia and distinctive facies, Aggrecan-type Spondyloepimetaphyseal Dysplasia and several idiopathic short stature phenotypes [8].
The height of an individual is a quantitative and multifactorial trait [9,10], where there is a genetic component related to the height of the parents that will determine a result equivalent to the average height of the parents, but its study is complex and necessarily involves diagnostic algorithms in which many factors other than genetic ones must be taken into account, including nutritional, endocrinological and environmental factors among others [11].
Among other aspects, these relevant clinical features to consider include the symmetry and proportion of the individual's body segments, and when the disproportion of these body segments can be evidenced accompanying a short stature, it is quite likely that we are facing a possible diagnosis of a skeletal dysplasia, which have different degrees of severity in terms of their manifestations and morbidity and mortality. Most skeletal dysplasias are of genetic origin and, therefore, together with a thorough clinical evaluation, including radiographic and other important data, molecular genetic study becomes a necessity to reach a definitive diagnosis [11].
Spondyloepiphyseal Dysplasia Kimberley type (SEDK), associated with the candidate gene by genetic linkage for the first time in 2002 in a South African family, is an autosomal dominant condition characterized by short proportionate stature, short trunk and limbs and early-onset osteoarthritis [12]. and its name is associated with mainly affecting the epiphyses of the long bones and vertebrae. Other manifestations include stubby body habitus; at the vertebral level patients may present with anterior osteophytes, vertebral endplate irregularity, sclerotic vertebral bodies and platyspondyly; at the pelvic level, flattened femoral head epiphyses and at the extremity level early-onset progressive osteoarthropathy, genu varum or genu valgum [13].
In our case, a probably pathogenic variant (class 2 variant) was detected in the previously undescribed ACAN gene, originating a premature stop codon, which results in a nonsense-mediated-degradation (NMD) of the mRNA of the mutant allele and, therefore, a haploinsufficiency for aggrecan is presumed [14]. The clinical manifestations of our patient, apart from the short stature of - 5 SD, the main cause of consultation and concern, are relatively mild, the most striking features being a slightly prominent forehead and slightly varus lower extremities (Figure 2).
The father of the proband, in whom the same mutation was detected, has a height of 1.61 m and within our general population could pass for a person with a slightly short average height, but not excessively striking, within the possible spectrum of the population. The only data that a priori could have made us think of a case of familial skeletal dysplasia was not excessively serious and had to do with the history of a herniated disc surgery at the age of 43, and that could be related to the person's lifestyle or other non-genetic factors.
Regarding treatment for SEDK, given that these patients have advanced bone age and premature growth cessation, the combination of puberty blockade by Gonadotropin-releasing hormone (GnRH) and timely use of growth hormone (GH) has proven to be beneficial for patients with this condition, resulting in gains in final height [7].
Osteoarthritis (OA) is a major cause of disability and source of social cost in older adults. It is a degenerative disorder of the synovial joints with an estimated prevalence of 10 to 15 % of adults over 60 years of age. Aggrecan degeneration is considered to be involved in this very common condition as a central part of its pathophysiology [15] [16]. Although its understanding has improved in recent years, the mechanisms involved and why in patients with SEDK OA presents prematurely remain to be fully understood. Therefore, patients with this condition and others with these genetic variants are the subject of study to clarify part of these mechanisms and to be able to find innovative therapeutic solutions, probably by regenerative medicine with stem cells [17].
CONCLUSIONS
Molecular genetic tests become an indispensable ally to complete diagnostic algorithms in cases of short stature that are unexplainable after the determination of endocrinological, nutritional or other non-genetic factors, since they can reveal the presence of pathogenic variants in related genes, thus allowing the diagnosis of skeletal dysplasias.
This case report shows us that there is a spectrum of variability in the height of individuals, which, at first glance, does not suggest a diagnosis of this type, but which may be explanatory and underlying a short average height accompanied by a range of orthopedic problems, with a variable spectrum of manifestations observed in some patients.
This case also gives us another example of the importance of guiding the change of thinking in everyday medical practice. This should be directed towards the routine use of molecular diagnosis to clarify the complex clinical panorama. On this occasion, due to variants in a single molecule, at least four different and apparently unrelated clinical entities can arise, which are part of the Aggrecanopathies.
Likewise, in routine medical practice, we are constantly faced with seemingly different entities but that have a single molecule as a common denominator, as has already been demonstrated in many other cases.
The diagnosis of SEDK, based on alterations in Aggrecan, provides a source of knowledge to understand why accelerated cartilage degeneration occurs in these cases and thus find more efficient therapeutic methods to treat a condition with such a high incidence as it is Osteoarthritis (OA).
ACKNOWLEDGMENTS
Thanks to Dr. Enrique A. Jimenez MD, FACC, FSCAI, Interventional Cardiologist at Overton Brooks VA Medical Center, Shreveport, Louisiana; Associate Director of the Cardiology Residency Program at Louisiana State University in Shreveport, Louisiana, for his contributions.
Thanks to Dr. Ernest Jermin, MD, Medical Director of the Behavioral Health Center Florida at the Cleveland Clinic Indian River Hospital, for his contributions.